You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5783)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (890)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (72)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Tee Cobre So EpcDocument1 pageTee Cobre So EpcAngel Rojas FrancoNo ratings yet
- Expt 2 - Thevenin-NortonDocument10 pagesExpt 2 - Thevenin-NortonNitin KhetadeNo ratings yet
- GE - Mark V Turbine ControlDocument1 pageGE - Mark V Turbine ControlAdil Butt0% (1)
- EndPl MomConn LSDDocument54 pagesEndPl MomConn LSDTony RoseNo ratings yet
- Calculation Sheet: Perunding Nusajasa SDN BHDDocument7 pagesCalculation Sheet: Perunding Nusajasa SDN BHDSarahLukakuNo ratings yet
- AGC AGC-CHEMICALS - Effect of Impurities On Membrane Performance & Recovery of Performance-Flemion Seminar 2012 PDFDocument59 pagesAGC AGC-CHEMICALS - Effect of Impurities On Membrane Performance & Recovery of Performance-Flemion Seminar 2012 PDFTahir KhalidNo ratings yet
- HeatPipe WrapAroundDocument2 pagesHeatPipe WrapAroundJohnson ChuaNo ratings yet
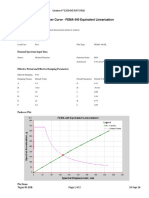
- FEMA 440 equivalent linearization pushover analysisDocument2 pagesFEMA 440 equivalent linearization pushover analysisAdam JrNo ratings yet
- Demand, Diversity & Utilization Factors for Electrical Load CalculationsDocument5 pagesDemand, Diversity & Utilization Factors for Electrical Load CalculationsJanitha HettiarachchiNo ratings yet
- Microsoft Word - Dangerous Goods Declaration PDFDocument1 pageMicrosoft Word - Dangerous Goods Declaration PDFAlfian AnasNo ratings yet
- Batchflux 5015 C: Electromagnetic Flowmeter For Volumetric Filling and BatchingDocument8 pagesBatchflux 5015 C: Electromagnetic Flowmeter For Volumetric Filling and BatchingavikbhaiNo ratings yet
- Lecture Notes 8b - Slope Deflection Frames No Sidesway PDFDocument12 pagesLecture Notes 8b - Slope Deflection Frames No Sidesway PDFShavin ChandNo ratings yet
- CIP CalculationDocument6 pagesCIP CalculationAnonymous fzP6QHQ100% (2)
- Elevator Inspections FINALDocument17 pagesElevator Inspections FINALrkarlinNo ratings yet
- 2GM (Mmbta56)Document3 pages2GM (Mmbta56)josealcantarafranco100% (1)
- Systemd in Suse Linux Enterprise 12 White PaperDocument16 pagesSystemd in Suse Linux Enterprise 12 White PaperhazardhapNo ratings yet
- XPol 806-960MHz 90° Fixed Tilt Sector Panel AntennaDocument1 pageXPol 806-960MHz 90° Fixed Tilt Sector Panel AntennaArif Noviardiansyah100% (1)
- Elma Combitest 419 Multifunctional Meter SpecificationsDocument4 pagesElma Combitest 419 Multifunctional Meter SpecificationscipriancordeaNo ratings yet
- Price ListDocument141 pagesPrice ListAriane Llantero0% (1)
- AdaptTo2016 Unit Testing With Sling and AEM Mocks Stefan SeifertDocument32 pagesAdaptTo2016 Unit Testing With Sling and AEM Mocks Stefan SeifertkaranNo ratings yet
- Palu Mallet KrisbowDocument1 pagePalu Mallet Krisbowhamidin_syarifNo ratings yet
- Earthquake Resistant Design PDFDocument5 pagesEarthquake Resistant Design PDFgavisita123No ratings yet
- GeForce RTX 3090 GA102 GF PG136-A03 Rev BDocument67 pagesGeForce RTX 3090 GA102 GF PG136-A03 Rev BSe SavNo ratings yet
- Avk Gate Valve, Flanged, Pn10/16 02/60-0045: EN 558-2 S.15/DIN F5, Clockwise To OpenDocument3 pagesAvk Gate Valve, Flanged, Pn10/16 02/60-0045: EN 558-2 S.15/DIN F5, Clockwise To OpenMohamed SayedNo ratings yet
- Math4220 PDFDocument10 pagesMath4220 PDFjhonmt7No ratings yet
- Aluminide and Silicide Diffusion Coatings by Pack Cementation For Nb-Ti-AlalloyDocument12 pagesAluminide and Silicide Diffusion Coatings by Pack Cementation For Nb-Ti-Alalloybrunoab89No ratings yet
- Steam Sterilization PrinciplesDocument8 pagesSteam Sterilization PrinciplesBhavik Thakar100% (1)
- It 504 A Artificial Intelligence Dec 2020Document4 pagesIt 504 A Artificial Intelligence Dec 2020ABHIMAT PANDEYNo ratings yet
- Principles of Operating SystemDocument127 pagesPrinciples of Operating Systempriyanka100% (1)
- Restricting TECO revoking after order settlementDocument4 pagesRestricting TECO revoking after order settlementMayank MevchaNo ratings yet