You might also like
- NOM-253-SSA1-2012 para La Disposicion de Sangre Humana y Sus Componentes Con Fines TerapeuticosDocument112 pagesNOM-253-SSA1-2012 para La Disposicion de Sangre Humana y Sus Componentes Con Fines Terapeuticossom1990No ratings yet
- Cartel FinalDocument1 pageCartel FinalAlejandro de la RosaNo ratings yet
- Protocolo en ExtensoDocument20 pagesProtocolo en ExtensoAlejandro de la RosaNo ratings yet
- PAHO Guia Leishmaniasis Americas 2013 Spa PDFDocument60 pagesPAHO Guia Leishmaniasis Americas 2013 Spa PDFMario Stiven Guambo MoranNo ratings yet
- Citopenias en LES PDFDocument9 pagesCitopenias en LES PDFAlejandro de la RosaNo ratings yet
- Guía de Clasificación Teratogénica de La FDA.Document15 pagesGuía de Clasificación Teratogénica de La FDA.Diana Elena Lopez0% (1)
- FACSDocument18 pagesFACSAbenamar Mejia PérezNo ratings yet
- Casos Clinicos HematologiaDocument70 pagesCasos Clinicos Hematologiacharly476100% (12)
- POZO 3er Examen Trabajo MensualDocument6 pagesPOZO 3er Examen Trabajo MensualAlejandro de la RosaNo ratings yet
- Revista Argentina De: NeurocirugíaDocument80 pagesRevista Argentina De: NeurocirugíaJacqueline AcurioNo ratings yet
- 08 15 Lovecocina PDFDocument100 pages08 15 Lovecocina PDFJavier100% (1)
- 2.5 Patologías Asociadas Al Metabolismo de LípidosDocument67 pages2.5 Patologías Asociadas Al Metabolismo de LípidosPaula Gil AlvarezNo ratings yet
- Laboratorio 1 Anatomía ElectroencefalografíaDocument13 pagesLaboratorio 1 Anatomía ElectroencefalografíaBRAYAN STEVEN CAICEDO CAICEDONo ratings yet
- Hechizos Treinta Hierbas Que CuranDocument10 pagesHechizos Treinta Hierbas Que CuranAzul Abat100% (1)
- Entrevistas Realizadas en CampoDocument9 pagesEntrevistas Realizadas en CampoJQNo ratings yet
- Farmacología Seminario 1Document3 pagesFarmacología Seminario 1Samer BastNo ratings yet
- Semana de Prevención 2023 - IIDocument19 pagesSemana de Prevención 2023 - IIMatita AlmaNo ratings yet
- Alimento de Bebe 6 MesesDocument9 pagesAlimento de Bebe 6 MesesJosé Luis RaymundoNo ratings yet
- Ensayo Simce Ciencias Naturales #3 SolucionarioDocument12 pagesEnsayo Simce Ciencias Naturales #3 SolucionarioCristian RomanNo ratings yet
- RV 7Document38 pagesRV 7ameNo ratings yet
- Codificacion Cie 10Document44 pagesCodificacion Cie 10TANIANo ratings yet
- 06 Hiperplasia Prostática Benigna - Dr. FloresDocument26 pages06 Hiperplasia Prostática Benigna - Dr. FloresNiquita19No ratings yet
- Hormona Gonadotropina Corionica HumanaDocument5 pagesHormona Gonadotropina Corionica HumanaJossNo ratings yet
- Defectos Anatómicos en El Camélido Sudamericano DoméDocument4 pagesDefectos Anatómicos en El Camélido Sudamericano DoméKdma DarkNo ratings yet
- Tesis Romain PDFDocument54 pagesTesis Romain PDFJP VdlcNo ratings yet
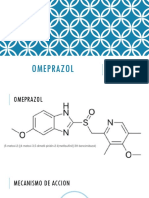
- Omeprazol ExposicionDocument10 pagesOmeprazol Exposicion徳利雅No ratings yet
- Manual de Procedimiento de EnfermeríaDocument72 pagesManual de Procedimiento de EnfermeríaWilliams David Ricra PariNo ratings yet
- Colico Renal, Caso ClínicoDocument7 pagesColico Renal, Caso Clínicoww.pacourgencias.blogspot.comNo ratings yet
- Micron Ebu Liz AdoresDocument2 pagesMicron Ebu Liz AdoresRAFAELNo ratings yet
- ODI Auxiliar de Servicios MenoresDocument7 pagesODI Auxiliar de Servicios MenoresJessica Evelyn Vidal OpazoNo ratings yet
- Medicina Forense AsfixiologíaDocument21 pagesMedicina Forense AsfixiologíaMilly HernándezNo ratings yet
- Dermatosis Producida Por Piojos Cear Omar Martinez Payan Gpo 7Document4 pagesDermatosis Producida Por Piojos Cear Omar Martinez Payan Gpo 7Alan MartinezNo ratings yet
- Cuadernillo de Trabajo 12 - 16 OctDocument5 pagesCuadernillo de Trabajo 12 - 16 OctMia SeremNo ratings yet
- Anemias Hemoliticas: Facultad de Ciencias Médicas Carrera de Laboratorio Clínico Msc. Mercedes TapiaDocument42 pagesAnemias Hemoliticas: Facultad de Ciencias Médicas Carrera de Laboratorio Clínico Msc. Mercedes TapiaKeicy Tehany Macias TorresNo ratings yet
- LECTURAS PARA ELABORAR Mapa ConceptualDocument2 pagesLECTURAS PARA ELABORAR Mapa Conceptualadrian vargasNo ratings yet
- Dermatitis Por Contacto AlergicaDocument17 pagesDermatitis Por Contacto AlergicaJosefina LomeliNo ratings yet
- Protocolo Único - Componente Práctico - Unidad 1 - Caso 3 - Componente Práctico - Protocolo LaboratorioDocument50 pagesProtocolo Único - Componente Práctico - Unidad 1 - Caso 3 - Componente Práctico - Protocolo LaboratorioNatalia Alfonso silvaNo ratings yet
- Perfil 20 NuevoDocument3 pagesPerfil 20 NuevoNestor SalgadoNo ratings yet
- Neuro CienciaDocument114 pagesNeuro CienciaJoseAlfaroDíazNo ratings yet