You might also like
- “Foundations to Flight: Mastering Physics from Curiosity to Confidence: Cipher 4”: “Foundations to Flight: Mastering Physics from Curiosity to Confidence, #4From Everand“Foundations to Flight: Mastering Physics from Curiosity to Confidence: Cipher 4”: “Foundations to Flight: Mastering Physics from Curiosity to Confidence, #4No ratings yet
- Chapter 3 ThermodynamicsDocument92 pagesChapter 3 ThermodynamicsRaymond KakalaNo ratings yet
- Chapter 03 Thermodynamics PDFDocument101 pagesChapter 03 Thermodynamics PDFPutri Nur Aisyah Halmy AzamNo ratings yet
- Nfpa 80 - 2007Document6 pagesNfpa 80 - 2007Artur MkrtchyanNo ratings yet
- P 7.4 ThermochemistryDocument32 pagesP 7.4 ThermochemistryFelicia GunawanNo ratings yet
- CHM432 Chap 1Document57 pagesCHM432 Chap 1KhalijahNo ratings yet
- ThermodynamicsDocument82 pagesThermodynamicsmdnishathasan141No ratings yet
- THERMOCHEMISTRY Hand Outs 2023Document6 pagesTHERMOCHEMISTRY Hand Outs 2023Paul Willard GumapacNo ratings yet
- LESSON 3 Chemistry FuelDocument11 pagesLESSON 3 Chemistry Fuelsimonjohn spanglerNo ratings yet
- Thermochemistry Y11Document23 pagesThermochemistry Y11ZlureNo ratings yet
- Intro 1a ThermochemistryDocument50 pagesIntro 1a ThermochemistryFatin IziantiNo ratings yet
- Thermodynamics and ThermochemistryDocument21 pagesThermodynamics and ThermochemistryElaiza Angelene NacarioNo ratings yet
- Thermochemistry: LESSON 1 Energy Changes in Chemical ReactionsDocument7 pagesThermochemistry: LESSON 1 Energy Changes in Chemical ReactionsAlexandra minNo ratings yet
- Ch06 SlidesDocument73 pagesCh06 SlidesbeelzeburtonNo ratings yet
- Thermo Chemistry-01-TheoryDocument15 pagesThermo Chemistry-01-TheoryRaju SinghNo ratings yet
- 1422 Notes Full 2010Document365 pages1422 Notes Full 2010Sreedevi KrishnakumarNo ratings yet
- Energy Changes in Chemical ReactionsDocument32 pagesEnergy Changes in Chemical ReactionsRon allen ConconNo ratings yet
- Thermochemistry PDFDocument37 pagesThermochemistry PDFSiddharth DhurandharNo ratings yet
- Unit 4: Thermochemistry and Nuclear Chemistry: Initial FinalDocument21 pagesUnit 4: Thermochemistry and Nuclear Chemistry: Initial FinalPankaj KumarNo ratings yet
- Chapter 5studentDocument44 pagesChapter 5studentDaniel ButenskyNo ratings yet
- Energy Relationships in Chemical ReactionsDocument11 pagesEnergy Relationships in Chemical ReactionsDanise NicoleNo ratings yet
- Thermochemistry: Chemical EnergyDocument24 pagesThermochemistry: Chemical EnergygnpanagiotouNo ratings yet
- Energetics of Chemical ReactionsDocument58 pagesEnergetics of Chemical ReactionsBichitra GautamNo ratings yet
- The First Law of Thermodynamics, Chapter 2Document14 pagesThe First Law of Thermodynamics, Chapter 2Natalie GibsonNo ratings yet
- Thermochemistry Summary - Libre TextsDocument4 pagesThermochemistry Summary - Libre Textsmacky 2No ratings yet
- Chem2 Q3 Week 5 6Document6 pagesChem2 Q3 Week 5 6Gwyneth CataneNo ratings yet
- Chapter 6 Lecture NotesDocument10 pagesChapter 6 Lecture NotesAhmad KamalNo ratings yet
- CO3 ThermodynamicsDocument72 pagesCO3 ThermodynamicsRonna IturaldeNo ratings yet
- Thermo ChemistryDocument96 pagesThermo Chemistryggallardo32642No ratings yet
- Blackboard Notes Chapter 6Document5 pagesBlackboard Notes Chapter 6Bill CampbellNo ratings yet
- Energetics and ThermochemistryDocument60 pagesEnergetics and ThermochemistryIsadora ThibauNo ratings yet
- 3 1 EnthalpyDocument30 pages3 1 EnthalpymahmoudNo ratings yet
- Thermodynamics Enthalpy and Hess LawDocument52 pagesThermodynamics Enthalpy and Hess LawKaithlyn LandichoNo ratings yet
- Combustion of MethaneDocument5 pagesCombustion of MethaneMuhammad Farhan AfifuddinNo ratings yet
- Thermodynamics: Specific Learning Objective at The End of The Session The Student Should Be Able To ExplainDocument67 pagesThermodynamics: Specific Learning Objective at The End of The Session The Student Should Be Able To ExplainfitriNo ratings yet
- ThermochemistryDocument2 pagesThermochemistryJun Cueva Comeros100% (1)
- Thermochemistry Part 1 PDFDocument21 pagesThermochemistry Part 1 PDFTintin LevidaNo ratings yet
- For Thermodynamics - F-1Document57 pagesFor Thermodynamics - F-1harshitaaanmol23No ratings yet
- Study ChemDocument13 pagesStudy ChemJanthina Rose AusteroNo ratings yet
- 7 Chapter 6Document58 pages7 Chapter 6azizNo ratings yet
- 1422 Chapt 15 ThermodynamicsDocument40 pages1422 Chapt 15 Thermodynamicstomm01No ratings yet
- Which Area of Chemistry Is Concerned With The Topic?: THERMOCHEMISTRY: Energy Changes in Chemical ReactionsDocument19 pagesWhich Area of Chemistry Is Concerned With The Topic?: THERMOCHEMISTRY: Energy Changes in Chemical ReactionsSahada KanapiyaNo ratings yet
- Chap 1 Thermodynamic (150318)Document74 pagesChap 1 Thermodynamic (150318)nurul atikaNo ratings yet
- ThermochemistryDocument50 pagesThermochemistrythobyy100% (4)
- ThermoschemsitryDocument40 pagesThermoschemsitryHadeel IbrahimNo ratings yet
- Chapter 6. Thermochemistry: Energy Flow and Chemical Change (Pages 156-177)Document36 pagesChapter 6. Thermochemistry: Energy Flow and Chemical Change (Pages 156-177)Joe NasalitaNo ratings yet
- Chapter 4 ThermoDocument64 pagesChapter 4 ThermoGiang ĐinhNo ratings yet
- Mew 1031 CH14Document29 pagesMew 1031 CH14Jorge Leonardo BedoyaNo ratings yet
- 4 Lecture 1 Thermo 1Document11 pages4 Lecture 1 Thermo 1Ahmed Al-ayatNo ratings yet
- Ap Chemistry Unit 5 Notes - ThermodynamicsDocument6 pagesAp Chemistry Unit 5 Notes - Thermodynamicsapi-336799605No ratings yet
- Chapter 5 - Thermochemistry GC2Document44 pagesChapter 5 - Thermochemistry GC2helalaNo ratings yet
- CHAPTER 8 (References)Document10 pagesCHAPTER 8 (References)JeromeNo ratings yet
- Lecture Handouts-2 2Document35 pagesLecture Handouts-2 2Ibrahim HersiNo ratings yet
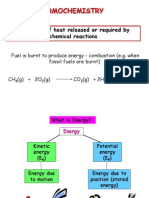
- Thermochemistry: The Study of Heat Released or Required by Chemical ReactionsDocument31 pagesThermochemistry: The Study of Heat Released or Required by Chemical ReactionsFifie Syfqh IINo ratings yet
- Thermochemistry: Ron RobertsonDocument21 pagesThermochemistry: Ron RobertsonRobin CelisNo ratings yet
- Enthalpy 1Document102 pagesEnthalpy 1kobegwapo595No ratings yet
- ThermochemistryDocument76 pagesThermochemistryChloe BascoNo ratings yet
- Chap 3 ThermochemistryDocument51 pagesChap 3 ThermochemistryChong RuYinNo ratings yet
- Chapter 6 - Thermochemistr - Part A FULLDocument19 pagesChapter 6 - Thermochemistr - Part A FULLPhillip WachowiakNo ratings yet
- Automation and Robotic Lec 2Document9 pagesAutomation and Robotic Lec 2Josh Sam Rindai MhlangaNo ratings yet
- MercerizationDocument141 pagesMercerizationkreeshnuNo ratings yet
- Lecture Plan Instructor K S RajmohanDocument4 pagesLecture Plan Instructor K S RajmohanSwapnil TripathiNo ratings yet
- Gem Lesson 1Document17 pagesGem Lesson 1Ritheria100% (1)
- Thermal Oil Boiler Vega PDFDocument2 pagesThermal Oil Boiler Vega PDFrafiradityaNo ratings yet
- Chap 9 Indices, Exponentials and Logarithms Part 1 PDFDocument44 pagesChap 9 Indices, Exponentials and Logarithms Part 1 PDFArahNo ratings yet
- Linear Response Theory: 3.1 Measurements and Correlation FunctionsDocument32 pagesLinear Response Theory: 3.1 Measurements and Correlation FunctionsJahan ClaesNo ratings yet
- Apa PR l238 I Joist - LPDocument14 pagesApa PR l238 I Joist - LPRodrigo CandeoNo ratings yet
- Chemistry Form 4 A NotesDocument67 pagesChemistry Form 4 A NotesJia En TanNo ratings yet
- Automatic Railway Gate Control by Using Microcontroller - 24 PagesDocument44 pagesAutomatic Railway Gate Control by Using Microcontroller - 24 PagesSebastin AshokNo ratings yet
- DIP Lecture 1&2 PDFDocument63 pagesDIP Lecture 1&2 PDFAhmedNo ratings yet
- Iso Dis 50006Document54 pagesIso Dis 50006abimanyubawono100% (3)
- Sade-5tnqzd r12 enDocument9 pagesSade-5tnqzd r12 enPop-Coman SimionNo ratings yet
- Institute of Seismological Research, Gandhinagar: "SAR Measurements For Earthquake Studies in India"Document16 pagesInstitute of Seismological Research, Gandhinagar: "SAR Measurements For Earthquake Studies in India"Santhosh Kumar BaswaNo ratings yet
- Ma8491 ModelDocument2 pagesMa8491 ModelANNo ratings yet
- Bearing Basics For Gas-Industry Screw CompressorsDocument5 pagesBearing Basics For Gas-Industry Screw CompressorsDayo IdowuNo ratings yet
- PPTC Operating Principle 11 04 2016 RaychemDocument20 pagesPPTC Operating Principle 11 04 2016 Raychemsvgl123No ratings yet
- Martini L4 TemperatureControlDocument11 pagesMartini L4 TemperatureControlJubaer JamiNo ratings yet
- SETTLING VELOCITY 2.1 - Calculations of Sedimentation Velocity and Hindered Settling Rate of ParticlesDocument74 pagesSETTLING VELOCITY 2.1 - Calculations of Sedimentation Velocity and Hindered Settling Rate of ParticlesSonu Singh100% (4)
- EPRI Cycle Chemistry Upsets During OperationDocument42 pagesEPRI Cycle Chemistry Upsets During OperationTrầnĐạtNo ratings yet
- Development of A Belt Conveyor For Small Scale Industry: September 2017Document6 pagesDevelopment of A Belt Conveyor For Small Scale Industry: September 2017DatNo ratings yet
- Air Compressor Summary ListDocument4 pagesAir Compressor Summary ListAlvin Smith100% (1)
- Experimental Physics PDFDocument2 pagesExperimental Physics PDFJessicaNo ratings yet
- Rieber Sealing in AmericaDocument10 pagesRieber Sealing in Americaulloap*100% (1)
- Trial Mix Design Report PDFDocument38 pagesTrial Mix Design Report PDFTimothy HughesNo ratings yet
- Bond Characteristics High-Strength Steel ReinforcementDocument6 pagesBond Characteristics High-Strength Steel ReinforcementUmair BaigNo ratings yet
- Critical Analysis of Properties of Ready Mix Concrete With Site Mix Concrete of Smart Road ProjectDocument6 pagesCritical Analysis of Properties of Ready Mix Concrete With Site Mix Concrete of Smart Road ProjectGolam Shahriar SakibNo ratings yet
- Technical DataDocument246 pagesTechnical DataABDUL GHAFOORNo ratings yet
- 50 Questions and Answers For Marine Engineers: Issue 3Document10 pages50 Questions and Answers For Marine Engineers: Issue 3Tara Gonzales100% (3)
- Ch4 Fluid KinematicsDocument30 pagesCh4 Fluid Kinematicsa u khan100% (1)