You might also like
- Curs 3 Lecture Nutrition and Bacteria CultivationDocument74 pagesCurs 3 Lecture Nutrition and Bacteria CultivationHåíthãm KhãtïßNo ratings yet
- Glycosides Classification GuideDocument28 pagesGlycosides Classification GuideApple Jane F. Bartolome100% (2)
- Reproduction and Growth in Bacteria: By: Hafiza Asfa Shafique Microbiology BS Biotechnology VDocument26 pagesReproduction and Growth in Bacteria: By: Hafiza Asfa Shafique Microbiology BS Biotechnology VJawadNo ratings yet
- The Cori Cycle PDFDocument1 pageThe Cori Cycle PDFvoon sjNo ratings yet
- Analytical - Exp 3 (Compile) DNSDocument11 pagesAnalytical - Exp 3 (Compile) DNSRemang GambangNo ratings yet
- Receptors As Drug Targets PDFDocument66 pagesReceptors As Drug Targets PDFAubreyNo ratings yet
- Semisolid Dosage Forms by BIBEK SINGH MAHATDocument105 pagesSemisolid Dosage Forms by BIBEK SINGH MAHATBibek Singh Mahat100% (1)
- Amino Acid MetabolismDocument15 pagesAmino Acid MetabolismMayur KaleNo ratings yet
- GLYCOSIDESDocument31 pagesGLYCOSIDESasma nizamNo ratings yet
- std12 Biochem em PDFDocument180 pagesstd12 Biochem em PDFPatil Jagadeesh ReddyNo ratings yet
- Macbeth Summery.Document2 pagesMacbeth Summery.Kafayetsaimum100% (1)
- Physical Pharmaceutics Û IIDocument2 pagesPhysical Pharmaceutics Û IIngel nilu80% (5)
- Practical Aspects of Good Pharmacy PracticeDocument26 pagesPractical Aspects of Good Pharmacy PracticeNur AjiNo ratings yet
- Routes of Drug EliminationDocument17 pagesRoutes of Drug EliminationdahiphalehNo ratings yet
- Bahauddin Zakariya University (BZU) Multan Subject List For BA Private CandidateDocument8 pagesBahauddin Zakariya University (BZU) Multan Subject List For BA Private CandidateAli ShahidNo ratings yet
- Lehninger Principles of Biochemistry PPT - Glycolysis For BBLDocument30 pagesLehninger Principles of Biochemistry PPT - Glycolysis For BBLJune HanNo ratings yet
- Drug MetabolismDocument78 pagesDrug Metabolismjuz gonzagaNo ratings yet
- THESISDocument41 pagesTHESISOlivia RamosNo ratings yet
- Biopharmaceutics and Pharmacokinetics in Drug ResearchDocument20 pagesBiopharmaceutics and Pharmacokinetics in Drug Researchlenanazarova1969No ratings yet
- Course No. Course Name Credits: Medicinal ChemistryDocument10 pagesCourse No. Course Name Credits: Medicinal ChemistryHeena BhojwaniNo ratings yet
- Combinatorial Chemistry: Dr. Nilesh PatelDocument21 pagesCombinatorial Chemistry: Dr. Nilesh PatelDr Nilesh PatelNo ratings yet
- Gatwech Dech RutDocument16 pagesGatwech Dech RutGatwech DechNo ratings yet
- Studies On The Pharmacokinetics and Pharmacodynamics of Mirtazapine in Healthy Young CatsDocument9 pagesStudies On The Pharmacokinetics and Pharmacodynamics of Mirtazapine in Healthy Young CatsWilliam ChandlerNo ratings yet
- Phenolic Profile and Antioxidant Activity of A Sempervivum Ruthenicum Koch Ethanolic ExtractDocument6 pagesPhenolic Profile and Antioxidant Activity of A Sempervivum Ruthenicum Koch Ethanolic ExtractNordsci ConferenceNo ratings yet
- Factors Influencing Drug Absorption Though Git PDFDocument59 pagesFactors Influencing Drug Absorption Though Git PDFRamakant JoshiNo ratings yet
- Descarte's Dream ArgumentDocument5 pagesDescarte's Dream ArgumentAlexander HenniesNo ratings yet
- Notes-Unit 3 - Instrumental Methods of AnalysisDocument40 pagesNotes-Unit 3 - Instrumental Methods of AnalysisAlexis UthaNo ratings yet
- Pharmacology overview and key conceptsDocument16 pagesPharmacology overview and key conceptscuolyNo ratings yet
- Major pathways of carbohydrate metabolismDocument119 pagesMajor pathways of carbohydrate metabolismsimasNo ratings yet
- Medicinal Chemistry Unit I IntroductionDocument27 pagesMedicinal Chemistry Unit I IntroductionjalilaNo ratings yet
- Poc II NotesDocument121 pagesPoc II NotesPonnam Chiranjeevi ChowdaryNo ratings yet
- Expt6 Sythesis of Phenacetin W15Document9 pagesExpt6 Sythesis of Phenacetin W15johnNo ratings yet
- Flavanoids, L 3Document48 pagesFlavanoids, L 3Ammadazfar ImamNo ratings yet
- Oscillometry and Conductometry: International Series of Monographs on Analytical ChemistryFrom EverandOscillometry and Conductometry: International Series of Monographs on Analytical ChemistryNo ratings yet
- Lesson 4.2. GlycoconjugatesDocument4 pagesLesson 4.2. GlycoconjugatesGemma CabañasNo ratings yet
- OintmentDocument12 pagesOintmentErfaneh FNNo ratings yet
- 2 CarbohydratesDocument49 pages2 CarbohydratesVishwanath SinduvadiNo ratings yet
- Slide Drug DeliveryDocument16 pagesSlide Drug DeliveryatikahNo ratings yet
- Signs in Language." This Understanding of Meaning Corresponds To of A Word Is Its Use in The Language' (In Other Words, The Role A WordDocument14 pagesSigns in Language." This Understanding of Meaning Corresponds To of A Word Is Its Use in The Language' (In Other Words, The Role A WordCarolyn TalabocNo ratings yet
- Common and Chemical Names of Some CompoundsDocument27 pagesCommon and Chemical Names of Some CompoundsnavinkapilNo ratings yet
- 2017-18 T.Y.B.Sc. Chemistry PDFDocument47 pages2017-18 T.Y.B.Sc. Chemistry PDFAkshay Khambare0% (1)
- Catabolism of HemeDocument34 pagesCatabolism of HemeGlen Jacobs Sumadihardja100% (2)
- Medicinal Chemistry Unit III Cholenergic Anti ChokenergicDocument32 pagesMedicinal Chemistry Unit III Cholenergic Anti ChokenergicjalilaNo ratings yet
- AntioxidAnt and AntimicrobiAl Activities of Leontice Eversmannii Roots. (MIC, DDPH, and ANTIOXIDANTS)Document10 pagesAntioxidAnt and AntimicrobiAl Activities of Leontice Eversmannii Roots. (MIC, DDPH, and ANTIOXIDANTS)CH NomiNo ratings yet
- 1-Biochemistry Chapter No 1Document63 pages1-Biochemistry Chapter No 1Mahrukh SaeedNo ratings yet
- Session 1 Introduction To PICS GMP 009-14Document12 pagesSession 1 Introduction To PICS GMP 009-14Elton SubijanoNo ratings yet
- Electrogravimetry - CHM4112LDocument7 pagesElectrogravimetry - CHM4112Lmarcia1416No ratings yet
- S.A. Raja Pharmacy College: Vi - Semester - (Iii-B.Pharm)Document51 pagesS.A. Raja Pharmacy College: Vi - Semester - (Iii-B.Pharm)MayurNo ratings yet
- Novel Drug Delivery SystemDocument5 pagesNovel Drug Delivery SystemshrikantthakurNo ratings yet
- Renal PharmacokineticsDocument20 pagesRenal PharmacokineticsChristopher VũNo ratings yet
- Organic Chemistry AUDocument91 pagesOrganic Chemistry AUAshley DayagNo ratings yet
- Alkaloids Introduction LectureDocument35 pagesAlkaloids Introduction LectureDaiene Paula100% (1)
- Digestive System NotesDocument10 pagesDigestive System NotesArchanna VyassNo ratings yet
- Drug Excretion-August 2020Document67 pagesDrug Excretion-August 2020Mussa Mwaitolage100% (1)
- Tablets PreformulationDocument90 pagesTablets Preformulationneha_dand1591No ratings yet
- AgentsDocument21 pagesAgentsmeomeogaugau26No ratings yet
- Combinatorial Chemistry: Guided By: Mr. R.T.LohiyaDocument23 pagesCombinatorial Chemistry: Guided By: Mr. R.T.LohiyaRinta MoonNo ratings yet
- AA Metabolism HANDOUTDocument4 pagesAA Metabolism HANDOUTnajwa hajjaliNo ratings yet
- Metabolism OF Amino Acids: Yulia SuciatiDocument37 pagesMetabolism OF Amino Acids: Yulia SuciatiAfnan AlkaffNo ratings yet
- Amino Acid MetabolismDocument25 pagesAmino Acid MetabolismParixit BhandurgeNo ratings yet
- Ex5 Milling MCDocument5 pagesEx5 Milling MCMuhammad RazaNo ratings yet
- 5 Microbiology ManualDocument59 pages5 Microbiology ManualMuhammad RazaNo ratings yet
- 5 Microbiology ManualDocument59 pages5 Microbiology ManualMuhammad RazaNo ratings yet
- 02 EL Bus Comm CH 02Document29 pages02 EL Bus Comm CH 02Muhammad RazaNo ratings yet
- DR Okunowo Wahab's Introductory Molecular Biology Lecture Note IIDocument39 pagesDR Okunowo Wahab's Introductory Molecular Biology Lecture Note IImodelprof100% (2)
- Fatty AcidDocument2 pagesFatty AcidFahad AhmedNo ratings yet
- PCR Primers Guide: Selecting Optimal PrimersDocument6 pagesPCR Primers Guide: Selecting Optimal PrimersDwi Novia PutriNo ratings yet
- Chapter 5 Biochemistry and Clinical Pathology Complete Notes by Noteskarts Acc To ER20Document5 pagesChapter 5 Biochemistry and Clinical Pathology Complete Notes by Noteskarts Acc To ER20prat.medbooksNo ratings yet
- Carboxylic Acid NotesDocument19 pagesCarboxylic Acid NotesGaurav KuntalNo ratings yet
- Exm 2014Document12 pagesExm 2014api-292477453No ratings yet
- Biology Cheat Sheet For EllaDocument3 pagesBiology Cheat Sheet For EllaJay EdwardsNo ratings yet
- DNA ReplicationDocument3 pagesDNA ReplicationChenra Missei YoroNo ratings yet
- Regulation of Plant Gene Expression by Antisense RNADocument28 pagesRegulation of Plant Gene Expression by Antisense RNAProject ICTNo ratings yet
- Q3-W4B-Protein Synthesis (By Group)Document5 pagesQ3-W4B-Protein Synthesis (By Group)Catherine CantadaNo ratings yet
- Grade 10 Science Module 2 Review on Transcription and TranslationDocument2 pagesGrade 10 Science Module 2 Review on Transcription and TranslationLanie De la TorreNo ratings yet
- IOFI Global Reference List CDS 8 December 2021Document75 pagesIOFI Global Reference List CDS 8 December 2021Brad HruskaNo ratings yet
- DNA and RNA Structure ComparisonDocument4 pagesDNA and RNA Structure ComparisonEdzborbonNo ratings yet
- DNA Replication in Prokaryotes ProjectDocument1 pageDNA Replication in Prokaryotes ProjectCliff LigulfNo ratings yet
- Amino Acid MCQ with AnswersDocument4 pagesAmino Acid MCQ with AnswersPrince AsanteNo ratings yet
- Biochemistry Midterm Quiz #1 - Module 4 QuestionsDocument1 pageBiochemistry Midterm Quiz #1 - Module 4 QuestionsLindsay SwanepeolNo ratings yet
- Replikasi DNA Dan Sintesis Protein (Gabungan)Document62 pagesReplikasi DNA Dan Sintesis Protein (Gabungan)adinda tyasprabandariNo ratings yet
- 5384 PDFDocument5 pages5384 PDFAngeline AguyenNo ratings yet
- Protein Synthesis - WorksheetDocument7 pagesProtein Synthesis - WorksheetMaisha IslamNo ratings yet
- Genetic CodeDocument16 pagesGenetic CodeSivagami Satish kumarNo ratings yet
- Biomacromolecules (Carbohydrate, Protein, Lipids, Minerals, Nucleic Acid)Document42 pagesBiomacromolecules (Carbohydrate, Protein, Lipids, Minerals, Nucleic Acid)Ram Kewal Tripathi100% (1)
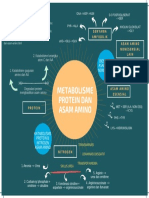
- Metabolisme Protein Dan Asam Amino: Mind Map TM Ii/Idk IDocument1 pageMetabolisme Protein Dan Asam Amino: Mind Map TM Ii/Idk ISania qurrataNo ratings yet
- Chapter 8Document28 pagesChapter 8Anupa GhoseNo ratings yet
- Fontenete2015 PDFDocument12 pagesFontenete2015 PDFManoMansoorNo ratings yet
- Soal BimbelDocument3 pagesSoal BimbelAndi Susan Dewi FNo ratings yet
- Nucleic Acid - An Overview - ScienceDirect TopicsDocument10 pagesNucleic Acid - An Overview - ScienceDirect TopicsBauza, Stephanie Dawn T.No ratings yet
- DNA StructureDocument31 pagesDNA Structuremohammed aliNo ratings yet
- Transcription and Translation: For Campbell Biology, Ninth EditionDocument85 pagesTranscription and Translation: For Campbell Biology, Ninth EditionClydeNo ratings yet
- Dha Algae Coa+Flow+HalalDocument5 pagesDha Algae Coa+Flow+HalalnadyaNo ratings yet
- DNA StructureDocument15 pagesDNA StructureOhhh OkayNo ratings yet