You might also like
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Solvay Group Presentation: Sinergia Cytec - Solvay: Una Historia de Innovacion, Sustentabilidad y LiderazgoDocument20 pagesSolvay Group Presentation: Sinergia Cytec - Solvay: Una Historia de Innovacion, Sustentabilidad y LiderazgoDaniel Calisaya AzpilcuetaNo ratings yet
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Wizard Genomic DNA Purification Kit ProtocolDocument21 pagesWizard Genomic DNA Purification Kit ProtocolDaniel Calisaya AzpilcuetaNo ratings yet
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (890)
- Overview of Generalized Disjunctive ProgrammingDocument51 pagesOverview of Generalized Disjunctive ProgrammingDaniel Calisaya AzpilcuetaNo ratings yet
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Optimization For Long Range Planning in The Chemical Industry PDFDocument15 pagesOptimization For Long Range Planning in The Chemical Industry PDFDaniel Calisaya AzpilcuetaNo ratings yet
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Tutorial BiopythonDocument61 pagesTutorial BiopythonDaniel Calisaya AzpilcuetaNo ratings yet
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- 375Document5 pages375Daniel Calisaya AzpilcuetaNo ratings yet
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
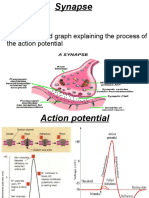
- Synapse Structure and Function ExplainedDocument11 pagesSynapse Structure and Function ExplainedMuhammad AbdullahNo ratings yet
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Dimauro 2017Document8 pagesDimauro 2017Eduardo RodriguezNo ratings yet
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Lab 1 - Introduction and ProtocolDocument28 pagesLab 1 - Introduction and ProtocolAnyaNo ratings yet
- J. Biol. Chem.-1988-Meister-17205-8Document4 pagesJ. Biol. Chem.-1988-Meister-17205-8Pepé DubNo ratings yet
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Drugs in The Pipeline For HBVDocument21 pagesDrugs in The Pipeline For HBVMarius StancuNo ratings yet
- Transudat Dan Eksudat by Dr. SidartiDocument15 pagesTransudat Dan Eksudat by Dr. SidartiEvelyn MaranantanNo ratings yet
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Protein Structure Powerpoint Presentation Newest Pink and PurpleDocument21 pagesProtein Structure Powerpoint Presentation Newest Pink and Purpleapi-281150432No ratings yet
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Prokaryotes vs Eukaryotes: Cell Structure and Key DifferencesDocument15 pagesProkaryotes vs Eukaryotes: Cell Structure and Key DifferencesGamu GamuNo ratings yet
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- Meiosis Notes MsDocument28 pagesMeiosis Notes Msapi-44119921No ratings yet
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Cell Respiration: ATP from GlucoseDocument11 pagesCell Respiration: ATP from GlucoseNiña Jennifer Potter Vergara100% (2)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- The Structure and Functions of the NucleusDocument19 pagesThe Structure and Functions of the NucleusYuu Ayu'k LifestarNo ratings yet
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- Anticoagulants, Fibrinolytics, AntiplateletsDocument88 pagesAnticoagulants, Fibrinolytics, Antiplateletspmuawiyah25No ratings yet
- Assessment of DNA Yield and Purity An Overlooked DDocument6 pagesAssessment of DNA Yield and Purity An Overlooked DKatrinaNo ratings yet
- ٣٢٥ Molecular biology Sabah Linjawi ١Document13 pages٣٢٥ Molecular biology Sabah Linjawi ١Zainab RaikNo ratings yet
- Genomics and Bioinformatics: Peter Gregory and Senthil NatesanDocument22 pagesGenomics and Bioinformatics: Peter Gregory and Senthil NatesanajaybioinfoNo ratings yet
- Importance of Biochemical Tests of BacteriaDocument5 pagesImportance of Biochemical Tests of Bacteriasandalaila100% (1)
- North Carolina Biology EOC Study GuideDocument38 pagesNorth Carolina Biology EOC Study GuideRaquel Galdamez-GaldamezNo ratings yet
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Gateway Cloning ManualDocument63 pagesGateway Cloning ManualdavidjkimNo ratings yet
- ANIMAL CELL-WPS OfficeDocument1 pageANIMAL CELL-WPS OfficeKelly Joem NievesNo ratings yet
- Biotechnology8 q1 Mod4 Biologicaltechniques v1-1Document30 pagesBiotechnology8 q1 Mod4 Biologicaltechniques v1-1Lleana PalesNo ratings yet
- DNA Restriction and Ligation in Gene Cloning, A Study Case in Prolidase Gene Cloning From Lactic Acid BacteriaDocument11 pagesDNA Restriction and Ligation in Gene Cloning, A Study Case in Prolidase Gene Cloning From Lactic Acid BacteriaRina ZaniaNo ratings yet
- Cloning Blunt-End DNA Fragments Into The Pgem - T Vector SystemsDocument5 pagesCloning Blunt-End DNA Fragments Into The Pgem - T Vector SystemsKhanh NhuNo ratings yet
- Chapter 5 The Structure and Function of Large Biological MoleculesDocument20 pagesChapter 5 The Structure and Function of Large Biological Molecules蔡旻珊No ratings yet
- Master of Science in Tropical and Infectious DiseasesDocument13 pagesMaster of Science in Tropical and Infectious DiseasesYalem ZewduNo ratings yet
- Solution Manual For Molecular Cell Biology 8th Edition Harvey Lodish Arnold Berk Chris A Kaiser Monty Krieger Anthony Bretscher Hidde Ploegh Angelika Amon Kelsey C Martin 33Document5 pagesSolution Manual For Molecular Cell Biology 8th Edition Harvey Lodish Arnold Berk Chris A Kaiser Monty Krieger Anthony Bretscher Hidde Ploegh Angelika Amon Kelsey C Martin 33PhilipWoodpsen100% (37)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Pengertian Dan Sejarah Bioteknologi - 2021Document39 pagesPengertian Dan Sejarah Bioteknologi - 2021Nila FirmaliaNo ratings yet
- Mutation PDFDocument11 pagesMutation PDFARCHANA BHARTI100% (1)
- Ex. 5 DNA Extraction PDFDocument3 pagesEx. 5 DNA Extraction PDFAlyssa Pauline PalacioNo ratings yet
- Vaccines 11 00206 v2Document31 pagesVaccines 11 00206 v2sesiaNo ratings yet
- Human Physiology From Cells To Systems 9th Edition Sherwood Test BankDocument26 pagesHuman Physiology From Cells To Systems 9th Edition Sherwood Test BankLauraMitchellfgie100% (55)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)