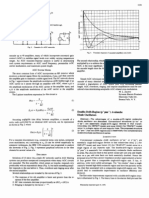
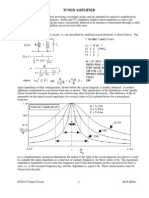
You might also like
- Pulses PDFDocument16 pagesPulses PDFAnonymous AFFiZnNo ratings yet
- Transformer Discharge DetectionDocument5 pagesTransformer Discharge DetectiondipenkhandhediyaNo ratings yet
- 35Cl and 14N Nuclear Quadrupole Resonance in Paradichlorobenzene, Sodium Chlorate, and HexamethylenetetramineDocument8 pages35Cl and 14N Nuclear Quadrupole Resonance in Paradichlorobenzene, Sodium Chlorate, and HexamethylenetetramineAllen MNo ratings yet
- 6oghz: Optical Millimetre-Wave Generation and Transmission Experiments For Mobile Band CommunicationsDocument3 pages6oghz: Optical Millimetre-Wave Generation and Transmission Experiments For Mobile Band CommunicationsMandeep SinghNo ratings yet
- PSK Demodulation (Part 1)Document10 pagesPSK Demodulation (Part 1)particlereddyNo ratings yet
- F. S. Tsung Et Al - Generation of Ultra-Intense Single-Cycle Laser Pulses by Using Photon DecelerationDocument4 pagesF. S. Tsung Et Al - Generation of Ultra-Intense Single-Cycle Laser Pulses by Using Photon DecelerationPocxaNo ratings yet
- Digital Communications Lab ExperimentsDocument0 pagesDigital Communications Lab Experimentsagama1188No ratings yet
- Performance Very Low Frequency Filters: Design Considerations For HighDocument4 pagesPerformance Very Low Frequency Filters: Design Considerations For Highmvmuthukumar88No ratings yet
- QPSK and 16qamDocument6 pagesQPSK and 16qamvelbon17006No ratings yet
- PAMDocument4 pagesPAMmukulgrd1No ratings yet
- D. Neely Et Al - Proposed Beatwave Experiment at RAL With The Vulcan CPA LaserDocument6 pagesD. Neely Et Al - Proposed Beatwave Experiment at RAL With The Vulcan CPA LaserDublin000No ratings yet
- Molecules 08 00092Document11 pagesMolecules 08 00092Thomas CharmNo ratings yet
- Sagnac Effect in Fiber GyroscopesDocument3 pagesSagnac Effect in Fiber GyroscopesRamiro Zakate ContrerasNo ratings yet
- Kaufmann 1976 Opt - Comm.17Document4 pagesKaufmann 1976 Opt - Comm.17Mark AldergroveNo ratings yet
- M. C. Downer Et Al - Plasma Channels and Laser Pulse Tailoring For GeV Laser-Plasma AcceleratorsDocument10 pagesM. C. Downer Et Al - Plasma Channels and Laser Pulse Tailoring For GeV Laser-Plasma AcceleratorsVasmazxNo ratings yet
- Pulsed-Field Gradients - Theory and Practice - Keeler, Clowes, Davis & LaueDocument63 pagesPulsed-Field Gradients - Theory and Practice - Keeler, Clowes, Davis & LaueChárbel FontesNo ratings yet
- RB Hyperfine StructureDocument3 pagesRB Hyperfine StructureKapila Wijayaratne100% (1)
- (1997) An Analysis of Two-Dimensional Pilot-Symbol Assisted Modulation For OFDMDocument4 pages(1997) An Analysis of Two-Dimensional Pilot-Symbol Assisted Modulation For OFDM賴勇先No ratings yet
- Direct RF Sampling Mixer With Recursive Filtering in Charge DomainDocument4 pagesDirect RF Sampling Mixer With Recursive Filtering in Charge DomainBinod AdhikariNo ratings yet
- Mod PamDocument7 pagesMod PamGanesh Kumar CNo ratings yet
- Gbit/s Direct Optical DPSK Modulation A 1530-nm DFB LaserDocument3 pagesGbit/s Direct Optical DPSK Modulation A 1530-nm DFB LaserKalyan TirunagariNo ratings yet
- Airborne Laser Scanning Basic Relations and Formulas 199-214Document16 pagesAirborne Laser Scanning Basic Relations and Formulas 199-214Grzegorz BrzęczyszczykiewiczNo ratings yet
- Ultra Narrow Band Modulation MethodsDocument14 pagesUltra Narrow Band Modulation MethodsAhmet SeçenNo ratings yet
- Of As: Double-Drift-Region (P ' P ') Diode OscillatorsDocument3 pagesOf As: Double-Drift-Region (P ' P ') Diode Oscillatorsanjan_debnathNo ratings yet
- Synchronization MethodsDocument7 pagesSynchronization MethodsAmit KumarNo ratings yet
- Precise Frequency Measurements of Methane Gas LinesDocument3 pagesPrecise Frequency Measurements of Methane Gas LinesThuanNo ratings yet
- Jresv67dn4p375 A1b PDFDocument7 pagesJresv67dn4p375 A1b PDFСаша СтојменовићNo ratings yet
- NARROW BAND AMPLIFIERSDocument8 pagesNARROW BAND AMPLIFIERSDeepraj BhujelNo ratings yet
- EEng 3210-ch3Document13 pagesEEng 3210-ch3Goitom HaileNo ratings yet
- D. O. Campos Et Al - The Application of A Multipass System For Thomson Scattering Diagnostics in Magnetically Con Ned PlasmasDocument5 pagesD. O. Campos Et Al - The Application of A Multipass System For Thomson Scattering Diagnostics in Magnetically Con Ned PlasmasMsdsxNo ratings yet
- 09 - Simultaneous Dual-Channel Stimulated Raman Scattering Microscopy Demultiplexed at Distinct Modulation FrequenciesDocument4 pages09 - Simultaneous Dual-Channel Stimulated Raman Scattering Microscopy Demultiplexed at Distinct Modulation Frequenciesyoussef ahmadNo ratings yet
- Blind Paper SpineDocument5 pagesBlind Paper SpineJoao PauloNo ratings yet
- Semiconductor Optical AmplifierDocument40 pagesSemiconductor Optical AmplifierVikas ThakurNo ratings yet
- Question BankDocument6 pagesQuestion Banksweetkhushboo786_592No ratings yet
- One-Pump Fiber Optical Parametric Amplifier inDocument6 pagesOne-Pump Fiber Optical Parametric Amplifier inRara Aisyah RamadhanyNo ratings yet
- Modulation: RZ-DQPSK Format Efficiency TowardsDocument2 pagesModulation: RZ-DQPSK Format Efficiency TowardsHank ChenNo ratings yet
- Plasma Diagnostics: Course Notes: Prof. F.F. Chen P A7: P DDocument23 pagesPlasma Diagnostics: Course Notes: Prof. F.F. Chen P A7: P Ddmp130No ratings yet
- PMD Tolerant Direct-Detection Optical OFDM SystemDocument2 pagesPMD Tolerant Direct-Detection Optical OFDM SystemZainab FaydhNo ratings yet
- Design of A Ring-Oscillator With A Wide Tuning Range in 0.13 M CMOS For The Use in Global Navigation Satellite SystemsDocument7 pagesDesign of A Ring-Oscillator With A Wide Tuning Range in 0.13 M CMOS For The Use in Global Navigation Satellite SystemsPaolo ColantonioNo ratings yet
- Frequency Reconfigurable Spirograph Planar Monopole Antenna (SPMA)Document4 pagesFrequency Reconfigurable Spirograph Planar Monopole Antenna (SPMA)usmanNo ratings yet
- ScienceDocument2 pagesScienceMike PoznerNo ratings yet
- 9.2 A 253mW/Channel 4TX/4RX Pulsed Chirping Phased-Array Radar TRX in 65nm CMOS For X-Band Synthetic - Aperture Radar ImagingDocument3 pages9.2 A 253mW/Channel 4TX/4RX Pulsed Chirping Phased-Array Radar TRX in 65nm CMOS For X-Band Synthetic - Aperture Radar ImagingBalaNo ratings yet
- Time-Division Multiplexing (TDM) Techniques and Line Coding MethodsDocument40 pagesTime-Division Multiplexing (TDM) Techniques and Line Coding Methodsmuhaned190No ratings yet
- VHF Radar Target Detection in The Presence of Clutter: Boriana VassilevaDocument8 pagesVHF Radar Target Detection in The Presence of Clutter: Boriana VassilevaGhada HamzaNo ratings yet
- Phase Shifter TutorialDocument12 pagesPhase Shifter TutorialsukusportyNo ratings yet
- Radar: Dept & Sem: Subject Name: Course Code: Unit: Prepared byDocument48 pagesRadar: Dept & Sem: Subject Name: Course Code: Unit: Prepared bySureshbabu PNo ratings yet
- Ren VLSI07Document2 pagesRen VLSI07jainarayanNo ratings yet
- Paper For The IMSDocument5 pagesPaper For The IMSmiguelgarciagarciaNo ratings yet
- Barov 2001 0120Document3 pagesBarov 2001 0120Particle Beam Physics LabNo ratings yet
- Paper Type: Whole Testpaper Test Date: 4 May 2008Document12 pagesPaper Type: Whole Testpaper Test Date: 4 May 2008Ayushi MaithaniNo ratings yet
- Lecture 8: Digital Modulation II: Chapter 5 - Modulation Techniques For Mobile RadioDocument63 pagesLecture 8: Digital Modulation II: Chapter 5 - Modulation Techniques For Mobile RadiobonfireeNo ratings yet
- University of Tripoli Faculty of Engineering Department of Electrical and Electronic EngineeringDocument8 pagesUniversity of Tripoli Faculty of Engineering Department of Electrical and Electronic EngineeringManal SalimNo ratings yet
- Ee3101 Lab Report ExpDocument16 pagesEe3101 Lab Report ExpyucesNo ratings yet
- Pulse ModulationDocument10 pagesPulse ModulationNitishNo ratings yet
- Semiclassical Four-Level Single-Atom LaserDocument4 pagesSemiclassical Four-Level Single-Atom LaserAnanta KusumaNo ratings yet
- Resonance Enhancement in Laser-Produced Plasmas: Concepts and ApplicationsFrom EverandResonance Enhancement in Laser-Produced Plasmas: Concepts and ApplicationsNo ratings yet
- Analysis and Design of Multicell DC/DC Converters Using Vectorized ModelsFrom EverandAnalysis and Design of Multicell DC/DC Converters Using Vectorized ModelsNo ratings yet
- Newnes Electronics Circuits Pocket Book (Linear IC): Newnes Electronics Circuits Pocket Book, Volume 1From EverandNewnes Electronics Circuits Pocket Book (Linear IC): Newnes Electronics Circuits Pocket Book, Volume 1Rating: 4.5 out of 5 stars4.5/5 (3)
- De HSG Lay Cau - Ban WordDocument124 pagesDe HSG Lay Cau - Ban WordNguyễn Hải YếnNo ratings yet
- Pengkondisian Kesiapan Belajar Untuk Pencapaian Hasil Belajar Dengan Gerakan Senam OtakDocument9 pagesPengkondisian Kesiapan Belajar Untuk Pencapaian Hasil Belajar Dengan Gerakan Senam OtakSaadah HasbyNo ratings yet
- High Yield Pics For STEP 2 CKDocument24 pagesHigh Yield Pics For STEP 2 CKKinan Alhalabi96% (28)
- Argenti, P. Corporate Communication. Cap. 8-9Document28 pagesArgenti, P. Corporate Communication. Cap. 8-9juan100% (1)
- List of SQAC DQAC SISC DISC 2019 20Document39 pagesList of SQAC DQAC SISC DISC 2019 20Shweta jainNo ratings yet
- Mohammad R. Mestarihi: About Me ObjectiveDocument1 pageMohammad R. Mestarihi: About Me ObjectiveMhmd MsttNo ratings yet
- 26 05 29 Hangers and Supports For Electrical SystemsDocument8 pages26 05 29 Hangers and Supports For Electrical SystemskaichosanNo ratings yet
- Polymer Science: Thermal Transitions in PolymersDocument20 pagesPolymer Science: Thermal Transitions in Polymerstanveer054No ratings yet
- Kashmira Karim Charaniya's ResumeDocument3 pagesKashmira Karim Charaniya's ResumeMegha JainNo ratings yet
- Impact of Endurance Exercise Training in the Fasted State on Muscle Metabolism and Insulin SensitivityDocument14 pagesImpact of Endurance Exercise Training in the Fasted State on Muscle Metabolism and Insulin SensitivityYo Vivo Fit Pablo y KarlaNo ratings yet
- BS EN 1677-5-2001 - Inc.Document3 pagesBS EN 1677-5-2001 - Inc.Ameer Sadimin SGNo ratings yet
- Albert PikeDocument6 pagesAlbert Pikeapi-302575383No ratings yet
- Intro - S4HANA - Using - Global - Bike - Slides - MM - en - v3.3 MODDocument45 pagesIntro - S4HANA - Using - Global - Bike - Slides - MM - en - v3.3 MODMrThedjalexNo ratings yet
- Differentiation SS2Document88 pagesDifferentiation SS2merezemenike272No ratings yet
- 60Hz Axial-Fan Centrifugal-Fan AC EN (2009) PDFDocument136 pages60Hz Axial-Fan Centrifugal-Fan AC EN (2009) PDFRodrigo GonçalvesNo ratings yet
- Why Leaders Should Look in the “MirrorDocument4 pagesWhy Leaders Should Look in the “MirrorCaryl Baylon EstreraNo ratings yet
- MARCOMDocument35 pagesMARCOMDrei SalNo ratings yet
- Transistor Amplifier Operating ParametersDocument21 pagesTransistor Amplifier Operating ParametersReddyvari VenugopalNo ratings yet
- EJC H2 Math P1 With Solution PDFDocument23 pagesEJC H2 Math P1 With Solution PDFKipp SohNo ratings yet
- Komposit UHMWPE Sebagai Alternatif Bantalan Rel Kereta Api: Abel Evan, Alia Kristika, Farid Mulia LatiefDocument11 pagesKomposit UHMWPE Sebagai Alternatif Bantalan Rel Kereta Api: Abel Evan, Alia Kristika, Farid Mulia LatiefAlia KristikaNo ratings yet
- Ubc 2015 May Sharpe JillianDocument65 pagesUbc 2015 May Sharpe JillianherzogNo ratings yet
- Johnson 1999Document20 pagesJohnson 1999Linh Hoàng PhươngNo ratings yet
- Analogue Lab Manual AL7212 V2.1-Panduan Praktek DSR Elektronika-DikonversiDocument235 pagesAnalogue Lab Manual AL7212 V2.1-Panduan Praktek DSR Elektronika-DikonversiAl-FarabiNo ratings yet
- EOD Stanags Overview April 2021Document12 pagesEOD Stanags Overview April 2021den mas paratate leo egoNo ratings yet
- We Generally View Objects As Either Moving or Not MovingDocument11 pagesWe Generally View Objects As Either Moving or Not MovingMarietoni D. QueseaNo ratings yet
- DelhiDocument40 pagesDelhiRahul DharNo ratings yet
- JA Ip42 Creating Maintenance PlansDocument8 pagesJA Ip42 Creating Maintenance PlansvikasbumcaNo ratings yet
- Fire Pump System Test ReportDocument12 pagesFire Pump System Test Reportcoolsummer1112143100% (2)
- Mpce 24Document39 pagesMpce 24Sachin Mehla0% (1)
- Educational Leadership Platform PaperDocument4 pagesEducational Leadership Platform Paperapi-273087939No ratings yet