You might also like
- Winmostar Tutorial: Gromacs Interfacial TensionDocument27 pagesWinmostar Tutorial: Gromacs Interfacial TensionEliasSMonteiroFilhoNo ratings yet
- Winmostar tutorial: Gromacs Vapor Pressure・Surface TensionDocument12 pagesWinmostar tutorial: Gromacs Vapor Pressure・Surface TensionEliasSMonteiroFilhoNo ratings yet
- Gromacs Tutorial 2 (Protein) PDFDocument19 pagesGromacs Tutorial 2 (Protein) PDFEliasSMonteiroFilhoNo ratings yet
- MD Gromacs TutorialDocument8 pagesMD Gromacs TutorialTAHRI DjillaliNo ratings yet
- Gromacs trajectory analysis library for flexible selectionsDocument13 pagesGromacs trajectory analysis library for flexible selectionsSimanta PaulNo ratings yet
- Fastran Tut 05 StagingDocument79 pagesFastran Tut 05 StagingYousaf SaidalaviNo ratings yet
- Orca Manual 3 0 3Document595 pagesOrca Manual 3 0 3Uallisson SantosNo ratings yet
- Winmostar tutorial: Gromacs Viscosity・Dielectric constantDocument12 pagesWinmostar tutorial: Gromacs Viscosity・Dielectric constantEliasSMonteiroFilhoNo ratings yet
- AutoDock Tutorial v1.2Document3 pagesAutoDock Tutorial v1.2Judith NelsonNo ratings yet
- DOCK6.1 TutorialDocument15 pagesDOCK6.1 Tutorial@lsreshtyNo ratings yet
- Auto Dock Steps in OrderDocument56 pagesAuto Dock Steps in Orderkishoreravi100% (1)
- MDWeb TutorialDocument26 pagesMDWeb TutorialpranavNo ratings yet
- QChem ManualDocument507 pagesQChem ManualLam KhoaNo ratings yet
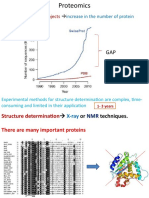
- Genome Sequencing Projects: Increase in The Number of Protein SequencesDocument27 pagesGenome Sequencing Projects: Increase in The Number of Protein SequencesBiju ThomasNo ratings yet
- Advance Thermodynamic Molecuar Dynamic Simulation: TA: Cuong LuuDocument24 pagesAdvance Thermodynamic Molecuar Dynamic Simulation: TA: Cuong LuuLuu Xuan CuongNo ratings yet
- Gromacs Tutorial For Drug Enzyme ComplexDocument21 pagesGromacs Tutorial For Drug Enzyme ComplexManoj Kumar RoutNo ratings yet
- Protein DockingDocument31 pagesProtein DockingFatima IffatNo ratings yet
- Welcome To Alke Ltd. Spiritual Arts and Science!Document9 pagesWelcome To Alke Ltd. Spiritual Arts and Science!thir20DNo ratings yet
- Pharmacophore-Based Molecular Docking: Bert E. Thomas, Diane Joseph-Mccarthy, Juan C.AvarezDocument45 pagesPharmacophore-Based Molecular Docking: Bert E. Thomas, Diane Joseph-Mccarthy, Juan C.Avarezsyahrul imranNo ratings yet
- Gromacs MD FlowchartDocument2 pagesGromacs MD FlowchartVivek SharmaNo ratings yet
- NTFK Revised 2012 - AmazonDocument230 pagesNTFK Revised 2012 - AmazonRuchika BakshiNo ratings yet
- Molecular Docking Using AutoDock and MGL Tools - GirinathDocument23 pagesMolecular Docking Using AutoDock and MGL Tools - GirinathGirinath Pillai60% (5)
- Prestressed I Section - IDEA StatiCaDocument12 pagesPrestressed I Section - IDEA StatiCaAlden Cayaga100% (1)
- PZ Cum User ManualDocument6 pagesPZ Cum User ManualxielfNo ratings yet
- 2D Adiabatic Compression Tutorial: Bottom Layering SetupDocument35 pages2D Adiabatic Compression Tutorial: Bottom Layering SetupmkbNo ratings yet
- 2D Adiabatic Compression TutorialDocument29 pages2D Adiabatic Compression Tutorialmm0hammadi100% (2)
- 2D Adiabatic Compression Layering TutorialDocument37 pages2D Adiabatic Compression Layering TutorialHoussam BEN SALAHNo ratings yet
- FLUENT - Tutorial - Dynamic Mesh - Missile Silo LaunchDocument30 pagesFLUENT - Tutorial - Dynamic Mesh - Missile Silo Launchmm0hammadiNo ratings yet
- Tutorial Ansys FluentDocument30 pagesTutorial Ansys FluentJulianLopezNo ratings yet
- Visual-Cast: Visual-Environment 13.5.2 Release Notes Release NotesDocument7 pagesVisual-Cast: Visual-Environment 13.5.2 Release Notes Release NotesMariano PinheiroNo ratings yet
- EECE 458: VLSI-II Sessional NAND Gate SimulationDocument10 pagesEECE 458: VLSI-II Sessional NAND Gate SimulationNishat Farha SharmiNo ratings yet
- Polyflow Extrusion WS06 Inverse ExtrusionDocument26 pagesPolyflow Extrusion WS06 Inverse ExtrusionTheerapat TaweebraksaNo ratings yet
- Sublevel Stope OptimazerDocument85 pagesSublevel Stope OptimazerNurlanOruziev100% (2)
- Place & Route Tutorial #1: I. SetupDocument13 pagesPlace & Route Tutorial #1: I. SetupDurgaPrasadNo ratings yet
- Vps 2008 Educ Example 1Document28 pagesVps 2008 Educ Example 1Charan KumarNo ratings yet
- RHSC Debug - For Internal Use Only - : 1 © 2018 ANSYS, Inc. ANSYS ConfidentialDocument76 pagesRHSC Debug - For Internal Use Only - : 1 © 2018 ANSYS, Inc. ANSYS Confidentialvaibhav_9090No ratings yet
- Fluent-Intro 17.0 WS06 Turbulent Flow Past A Backward Facing StepDocument32 pagesFluent-Intro 17.0 WS06 Turbulent Flow Past A Backward Facing StepFabiano Lebkuchen100% (1)
- Prestressed Saddle Beam - IDEA StatiCaDocument19 pagesPrestressed Saddle Beam - IDEA StatiCaAlden CayagaNo ratings yet
- User Manual For Frequently Used Property Spread Sheet - Yocho - v01 - s26 - TempDocument7 pagesUser Manual For Frequently Used Property Spread Sheet - Yocho - v01 - s26 - Tempchldudwo2158No ratings yet
- Exclusive DVD in Motion Activation For CIC by BMWCODING v2Document13 pagesExclusive DVD in Motion Activation For CIC by BMWCODING v2TimSmithNo ratings yet
- DVD in Motion Activation For CIC by BMWCODING v2Document13 pagesDVD in Motion Activation For CIC by BMWCODING v2Kunihei YanagawaNo ratings yet
- Place & Route Tutorial #1: I. SetupDocument13 pagesPlace & Route Tutorial #1: I. Setupijalab1No ratings yet
- Polyflow BMTF WS01 Thermoforming of A BlisterDocument34 pagesPolyflow BMTF WS01 Thermoforming of A Blisterwoongs73No ratings yet
- and Start Maestro On Your Laptop (Recommended)Document6 pagesand Start Maestro On Your Laptop (Recommended)Neil SNo ratings yet
- Fatigue Analysis (Damage Calculation) Using S-N ApproachDocument7 pagesFatigue Analysis (Damage Calculation) Using S-N ApproachdhanrajNo ratings yet
- Modeling Loads and Load Combinations - TRNC03271Document41 pagesModeling Loads and Load Combinations - TRNC03271Kidd TornoNo ratings yet
- Basics of Embedded Systems (RX63N) - ExercisesDocument27 pagesBasics of Embedded Systems (RX63N) - ExercisesWarenya RanminiNo ratings yet
- Practice Workbook: Designing Concrete Structures Using The Reinforced Concrete DesignerDocument47 pagesPractice Workbook: Designing Concrete Structures Using The Reinforced Concrete DesignerKidd TornoNo ratings yet
- Box Beam With Transient LoadDocument47 pagesBox Beam With Transient LoadManasses juniorNo ratings yet
- OptiStruct - 01 - Design Concept For A Structural C-ClipDocument12 pagesOptiStruct - 01 - Design Concept For A Structural C-ClipBaljinder SinghNo ratings yet
- Whittle Introductory Poly Metallic TutorialDocument21 pagesWhittle Introductory Poly Metallic Tutorialhenry_mineroNo ratings yet
- Tutorial 28 Coal Mine StopeDocument18 pagesTutorial 28 Coal Mine Stoperongow titoNo ratings yet
- XFEM Crack Propagation TutorialDocument19 pagesXFEM Crack Propagation TutorialVarghese MathewNo ratings yet
- SAP2000 Tutorial Example: Analysis and Design of Continuous RC BeamDocument21 pagesSAP2000 Tutorial Example: Analysis and Design of Continuous RC BeamafghankhanNo ratings yet
- Hints For Cadence VirtuosoDocument7 pagesHints For Cadence VirtuosoThomas GeorgeNo ratings yet
- NXU 2020 Hands On Activity Book UpdatedDocument43 pagesNXU 2020 Hands On Activity Book UpdatedHeribertoNo ratings yet
- Design of A Double Corbel Using CAST Per ACI 318-02 Appendix A, SI UnitDocument41 pagesDesign of A Double Corbel Using CAST Per ACI 318-02 Appendix A, SI Unityoga arkanNo ratings yet
- Fastran Tut 02 Naca0012Document29 pagesFastran Tut 02 Naca0012Yousaf SaidalaviNo ratings yet
- Reservoir Chracterization Lab Manual (PEC 506) Version 2Document98 pagesReservoir Chracterization Lab Manual (PEC 506) Version 2Yash GuptaNo ratings yet
- Modeling Loads in The SPPM - TRNC03078Document39 pagesModeling Loads in The SPPM - TRNC03078Kidd TornoNo ratings yet
- 6th Central Pay Commission Salary CalculatorDocument15 pages6th Central Pay Commission Salary Calculatorrakhonde100% (436)
- SM Chapter3 PDFDocument20 pagesSM Chapter3 PDFJuan perezNo ratings yet
- Dioxido de Titanio en Forma AnataseDocument8 pagesDioxido de Titanio en Forma AnataseJose PabloNo ratings yet
- SM Chapter1 PDFDocument14 pagesSM Chapter1 PDFgoc1794No ratings yet
- Classical Canonical TransfDocument8 pagesClassical Canonical TransfJose PabloNo ratings yet
- Setting vpn1Document10 pagesSetting vpn1Unink AanNo ratings yet
- SRSUNTOUR General Fork GlossaryDocument23 pagesSRSUNTOUR General Fork GlossaryThomas JunkersfeldNo ratings yet
- Studio GPGL LogDocument5 pagesStudio GPGL LogCarlos Julian LemusNo ratings yet
- Internet Controlled Multifunctional UGV For SurvellianceDocument74 pagesInternet Controlled Multifunctional UGV For SurvellianceMd Khaled NoorNo ratings yet
- The Sperry Bombsight: A Pioneering Rival to NordenDocument6 pagesThe Sperry Bombsight: A Pioneering Rival to NordenPaul DupontNo ratings yet
- Achievements Under EpiraDocument6 pagesAchievements Under EpiraLyn Dela Cruz DumoNo ratings yet
- Friction Factor For Turbulent Pipe Flow: January 2006Document17 pagesFriction Factor For Turbulent Pipe Flow: January 2006John AnthoniNo ratings yet
- Interaction & Bank EffectDocument6 pagesInteraction & Bank EffectAkash KandwalNo ratings yet
- Concrete: Concrete Is A Composite Material Composed of Fine and CoarseDocument36 pagesConcrete: Concrete Is A Composite Material Composed of Fine and CoarseclubmailusNo ratings yet
- The Causes and Prevention of Crowd DisastersDocument10 pagesThe Causes and Prevention of Crowd DisastersVarun SwaminathanNo ratings yet
- BTS Training Fiber Optic Advanced SplicingDocument5 pagesBTS Training Fiber Optic Advanced Splicingjama99No ratings yet
- DMD Documentation Error - Freetronics ForumDocument3 pagesDMD Documentation Error - Freetronics ForumapofviewNo ratings yet
- AASHTO T283-22 Standard Method of Test for Resistance of Compacted Asphalt Mixtures to Moisture-Induced DamageDocument11 pagesAASHTO T283-22 Standard Method of Test for Resistance of Compacted Asphalt Mixtures to Moisture-Induced DamageErnesto Oscar VidelaNo ratings yet
- Unit-I: Introduction To J2EEDocument29 pagesUnit-I: Introduction To J2EEsurakshaNo ratings yet
- Acsomega 9b01541Document9 pagesAcsomega 9b01541Benedictus EduardoNo ratings yet
- Operator Interface Hmi Touch Screen Cmore - 10Document54 pagesOperator Interface Hmi Touch Screen Cmore - 10QuantumAutomation100% (1)
- AS1895/7 E-FLEX Sealing Solutions: Part Number AS1895/7 Reference Duct Size Seal DimensionsDocument1 pageAS1895/7 E-FLEX Sealing Solutions: Part Number AS1895/7 Reference Duct Size Seal DimensionsAlex Zambrana RodríguezNo ratings yet
- Apache Oozie - A workflow scheduler to manage Hadoop jobsDocument5 pagesApache Oozie - A workflow scheduler to manage Hadoop jobsarjuncchaudharyNo ratings yet
- Manual Servicio SubaruDocument5,963 pagesManual Servicio SubaruCristian Mauricio Alarcon RojasNo ratings yet
- Sad Thesis Guidelines FinalsDocument13 pagesSad Thesis Guidelines FinalsJes RamosNo ratings yet
- Gps VulnerabilityDocument28 pagesGps VulnerabilityaxyyNo ratings yet
- Ice-Lined Refrigerator ManualDocument8 pagesIce-Lined Refrigerator ManualEmilioPerezBallesterosNo ratings yet
- Linear Slot DiffuserDocument15 pagesLinear Slot DiffuserhyderabadNo ratings yet
- GD&T Training Levels and ServicesDocument1 pageGD&T Training Levels and ServicesdramiltNo ratings yet
- AMS Thread Size ChartDocument4 pagesAMS Thread Size Chartarunvelu_1250% (2)
- JUS StandardsDocument28 pagesJUS StandardsgarejkaNo ratings yet
- Customer Targeted E-CommerceDocument4 pagesCustomer Targeted E-CommercepriyaNo ratings yet
- DiodeDocument22 pagesDiodeSaurabh Mittal100% (1)
- Munsell Color Charts and GaugesDocument2 pagesMunsell Color Charts and GaugesMario DalengkadeNo ratings yet