You might also like
- Apostila Completa de MetrologiaDocument94 pagesApostila Completa de Metrologiaaguiark510100% (7)
- Reinos de Ferro - Aventuras Urbanas DigitalDocument99 pagesReinos de Ferro - Aventuras Urbanas DigitalBrunoAlmeidadeLima100% (3)
- Cap 06 ReagentesDocument35 pagesCap 06 ReagentesvanessaNo ratings yet
- Usinagem Por EletroerosãoDocument30 pagesUsinagem Por EletroerosãoRafael CavalcantiNo ratings yet
- A Morte e Seu Mistério - (2) Durante A Morte - Camille FlammarionDocument325 pagesA Morte e Seu Mistério - (2) Durante A Morte - Camille Flammariontupan84No ratings yet
- A Morte e Seu Mistério - (2) Durante A Morte - Camille FlammarionDocument325 pagesA Morte e Seu Mistério - (2) Durante A Morte - Camille Flammariontupan84No ratings yet
- O Desconhecido e Os Problemas Psiquicos - Camille Flammarion PDFDocument552 pagesO Desconhecido e Os Problemas Psiquicos - Camille Flammarion PDFtiago_henrique_3No ratings yet
- Determinação de Cloro Residual e Da Demanda de Cloro - Portal TADocument4 pagesDeterminação de Cloro Residual e Da Demanda de Cloro - Portal TAJeová Dantas100% (1)
- Fabricação de Produtos Higiene PessoalDocument37 pagesFabricação de Produtos Higiene PessoalAntonio Ariza Neto ArizaNo ratings yet
- Lista de Exerccios 3respostas - Misturas e SoluesDocument1 pageLista de Exerccios 3respostas - Misturas e SoluesGabriel Brito PamplonaNo ratings yet
- Sabesp NTS 015 - NH3Document8 pagesSabesp NTS 015 - NH3604166No ratings yet
- BiomagnetismoDocument14 pagesBiomagnetismoShalom Adonai100% (1)
- Revisão Geral ArquivologiaDocument17 pagesRevisão Geral ArquivologiaLucas SadeckNo ratings yet
- Ligas MetálicasDocument34 pagesLigas MetálicasSillvano PiresNo ratings yet
- Embrapa - Vegetais em ConservaDocument48 pagesEmbrapa - Vegetais em ConservaSergio Fagundes100% (1)
- FA Nº4 - 7ano - 19 20Document8 pagesFA Nº4 - 7ano - 19 20Cristina RodriguesNo ratings yet
- A Morte e Seu Mistério - (1) Antes Da Morte - Camille FlammarionDocument386 pagesA Morte e Seu Mistério - (1) Antes Da Morte - Camille Flammariontupan84No ratings yet
- Sulfito - Ondeo NalcoDocument4 pagesSulfito - Ondeo NalcoAndre Paz100% (1)
- Camille Flammarion - Estela PDFDocument299 pagesCamille Flammarion - Estela PDFMilton LeiteNo ratings yet
- A Morte e Seu Mistério - (3) Depois Da Morte - Camille FlammarionDocument389 pagesA Morte e Seu Mistério - (3) Depois Da Morte - Camille Flammariontupan84100% (1)
- Carefree - FISPQDocument13 pagesCarefree - FISPQMurilo Pucineli50% (2)
- NBR 12620 - Aguas - Determinacao de Nitrato - Metodos Do Acido Cromotropico e Do Acido FenoldissuDocument5 pagesNBR 12620 - Aguas - Determinacao de Nitrato - Metodos Do Acido Cromotropico e Do Acido FenoldissuCassiaSobanski100% (2)
- 4500-N A. IntroduçãoDocument34 pages4500-N A. IntroduçãoFábio Machry SanchesNo ratings yet
- Prática Sais de Diazonio CorantesDocument4 pagesPrática Sais de Diazonio CorantesEmilly SilvaNo ratings yet
- NBR 13802 (Abr 1997) - Água - Determinação de Selênio Pelo Método Colorimétrico Da DiaminobenzidinaDocument3 pagesNBR 13802 (Abr 1997) - Água - Determinação de Selênio Pelo Método Colorimétrico Da DiaminobenzidinaYuri Bahia de VasconcelosNo ratings yet
- 3-Fosfato Dissolv PDFDocument3 pages3-Fosfato Dissolv PDFSergio CrepaldiNo ratings yet
- 31 Demanda Química de Oxigênio DQODocument11 pages31 Demanda Química de Oxigênio DQOJoão Paulo Mendes FerreiraNo ratings yet
- Trabalho 2 de TeoqDocument15 pagesTrabalho 2 de TeoqThays SantosNo ratings yet
- MÉTODO DE PARARROSANILINA (Determinação SO2 No Ar)Document19 pagesMÉTODO DE PARARROSANILINA (Determinação SO2 No Ar)eamarNo ratings yet
- Nitrogenio PDFDocument5 pagesNitrogenio PDFPedro Paulo100% (1)
- NTS221 - Nitrogênio Amoniacal, Orgânico e Total Kjeldahl - Método TitulométricoDocument10 pagesNTS221 - Nitrogênio Amoniacal, Orgânico e Total Kjeldahl - Método TitulométricoPedro CarmonaNo ratings yet
- Monografias Do Volume 2 - Letra P PDFDocument57 pagesMonografias Do Volume 2 - Letra P PDFFrederico MacienteNo ratings yet
- Relatorio Da P-NitroacetanilidaDocument12 pagesRelatorio Da P-NitroacetanilidaEvaldo XimenesNo ratings yet
- DQO Refluxo FechadoDocument3 pagesDQO Refluxo FechadoRô BatistaNo ratings yet
- Analises de Aguas ResiduariasDocument39 pagesAnalises de Aguas ResiduariasJulia Kaiane PratesNo ratings yet
- Método para Determinação de Carbono Orgânico Do SoloDocument5 pagesMétodo para Determinação de Carbono Orgânico Do SoloLidiane Assis GuimaraesNo ratings yet
- Aplicaes de Volumetria de Oxi-ReduoDocument8 pagesAplicaes de Volumetria de Oxi-ReduoAna Myrta0% (1)
- NBR 13796 (Abr 1997) - Água - Determinação de Nitrogênio Orgânico, Kjeldahl e Total - Métodos Macro e Semimicro KjeldahlDocument5 pagesNBR 13796 (Abr 1997) - Água - Determinação de Nitrogênio Orgânico, Kjeldahl e Total - Métodos Macro e Semimicro KjeldahlYuri Bahia de Vasconcelos100% (3)
- Prática de CloretosDocument5 pagesPrática de CloretosMaria Isabel CabralNo ratings yet
- CLORO RESIDUAL LIVREv 05Document5 pagesCLORO RESIDUAL LIVREv 05bispojjosNo ratings yet
- NBR 10739 - 1989 - Agua - Determinacao de Oxigenio Consumido - Metodo Do Permanganato de PotassioDocument3 pagesNBR 10739 - 1989 - Agua - Determinacao de Oxigenio Consumido - Metodo Do Permanganato de PotassioItalo Lacerda Fernandes100% (3)
- Revisão Final EnemDocument21 pagesRevisão Final EnemFrancilio Santos da CruzNo ratings yet
- EXPERIMENTO - Determinação de Oxigênio Dissolvido - ODDocument3 pagesEXPERIMENTO - Determinação de Oxigênio Dissolvido - ODDjjota FloripaNo ratings yet
- Minicurso Preparo Padronizacao SolucoesDocument33 pagesMinicurso Preparo Padronizacao SolucoesJanaina LeitinhoNo ratings yet
- 2-Protocolo de DQO Refluxo AbertoDocument10 pages2-Protocolo de DQO Refluxo AbertoRené Villas Bôas Dos SantosNo ratings yet
- Determinação de CLORETOS EM ÁGUASDocument2 pagesDeterminação de CLORETOS EM ÁGUASdiangiquimicoNo ratings yet
- 2542 Aula 09 DBODocument2 pages2542 Aula 09 DBOAlexandre ItoNo ratings yet
- NBR 10740 - 1989 - Agua - Determinacao de Fenol TotalDocument6 pagesNBR 10740 - 1989 - Agua - Determinacao de Fenol TotalbillalvaroNo ratings yet
- 4 Od PDFDocument4 pages4 Od PDFSergio CrepaldiNo ratings yet
- NTS009Document10 pagesNTS009Lauren RothNo ratings yet
- DETERMINAÇÃO DE NITRITOS EM ÁGUAS - Química - UTFPR - 2010Document14 pagesDETERMINAÇÃO DE NITRITOS EM ÁGUAS - Química - UTFPR - 2010joaomarcoslsNo ratings yet
- NBR 12614 - Aguas - Determinacao Da Demanda Bioquimica de Oxigenio (DBO)Document5 pagesNBR 12614 - Aguas - Determinacao Da Demanda Bioquimica de Oxigenio (DBO)Raquel LimaNo ratings yet
- Procedimentos Tecnicos para Analise Fisico-Quimica Da AguaDocument21 pagesProcedimentos Tecnicos para Analise Fisico-Quimica Da Aguanigilberto100% (1)
- NBR 10559 - 1988 - Aguas - Determinacao de Oxigenio Dissolvido - Metodo Iodometrico de Winkler eDocument11 pagesNBR 10559 - 1988 - Aguas - Determinacao de Oxigenio Dissolvido - Metodo Iodometrico de Winkler eAudrey Xavier0% (1)
- Relatório - Oxigênio DissolvidoDocument10 pagesRelatório - Oxigênio DissolvidoRIAN CAMPOS ALMEIDANo ratings yet
- Determinação CloretosDocument4 pagesDeterminação CloretosPAOLA GRIEBELER FERREIRANo ratings yet
- Artigo Cientifico Cloro AtivoDocument4 pagesArtigo Cientifico Cloro Ativojackson gomesNo ratings yet
- Aula 6. Identificação e Quantificação de Produtos NaturaisDocument80 pagesAula 6. Identificação e Quantificação de Produtos NaturaisRose SchneiderNo ratings yet
- DETERMINAÇÃO VOLUMÉTRICA DE ÁCIDO ASCÓRBICO EM VITAMINA C MERCK CEBION® - Química - UTFPR - 2010Document12 pagesDETERMINAÇÃO VOLUMÉTRICA DE ÁCIDO ASCÓRBICO EM VITAMINA C MERCK CEBION® - Química - UTFPR - 2010joaomarcosls100% (6)
- Aula 02 - Tratamento de à - GuaDocument4 pagesAula 02 - Tratamento de à - GuaSUYANNE TESKE PIRESNo ratings yet
- Demanda Bioquímica de Oxigênio - Dbo5Document9 pagesDemanda Bioquímica de Oxigênio - Dbo5hanyelleNo ratings yet
- Metodo para Silica Reativa Ao MolibdatoDocument1 pageMetodo para Silica Reativa Ao MolibdatoastreasilvaNo ratings yet
- NBR 13797 (Abr 1997) - Água - Determinação de Cloretos - Métodos Titulométricos Do Nitrato Mercúrico e Do Nitrato de PrataDocument5 pagesNBR 13797 (Abr 1997) - Água - Determinação de Cloretos - Métodos Titulométricos Do Nitrato Mercúrico e Do Nitrato de PrataYuri Bahia de Vasconcelos100% (3)
- Exp - 9 - Síntese de Salicilato de MetilaDocument2 pagesExp - 9 - Síntese de Salicilato de MetilaLeili AlmeidaNo ratings yet
- 1 IntroduçãoDocument2 pages1 IntroduçãoEdson AlvesNo ratings yet
- 35 RifampicinaDocument6 pages35 RifampicinalucasNo ratings yet
- POP-FQ-02 - Determinação de SO2 Pelo Método Do H2O2Document9 pagesPOP-FQ-02 - Determinação de SO2 Pelo Método Do H2O2deyverson freitasNo ratings yet
- POP-LAB-012 Proc. Analítico - Índice de PeróxidoDocument3 pagesPOP-LAB-012 Proc. Analítico - Índice de PeróxidoVictor GabrielNo ratings yet
- 22 Super Benefícios do Bicarbonato de Sódio: O bicarbonato de sódio tem uma variedade de usos domésticos adicionais e benefícios à saúdeFrom Everand22 Super Benefícios do Bicarbonato de Sódio: O bicarbonato de sódio tem uma variedade de usos domésticos adicionais e benefícios à saúdeNo ratings yet
- Utilização do lodo gerado na ETA de Alvorada-RS na fabricação de blocos cerâmicosFrom EverandUtilização do lodo gerado na ETA de Alvorada-RS na fabricação de blocos cerâmicosNo ratings yet
- Biofísica para ciências biomédicas – 4ª ediçãoFrom EverandBiofísica para ciências biomédicas – 4ª ediçãoNo ratings yet
- Porto Belo SC Compilada (12 06 2019)Document56 pagesPorto Belo SC Compilada (12 06 2019)tiago_henrique_3No ratings yet
- 40 Roteiros Dos Videos Curso EnapDocument24 pages40 Roteiros Dos Videos Curso Enaptiago_henrique_3No ratings yet
- Lei Complementar 319Document25 pagesLei Complementar 319tiago_henrique_3No ratings yet
- Lei Complementar 326 2018Document7 pagesLei Complementar 326 2018tiago_henrique_3No ratings yet
- Lei Complementar 25 2013 Sao Miguel Do Oeste SC Compilada (25 09 2019)Document60 pagesLei Complementar 25 2013 Sao Miguel Do Oeste SC Compilada (25 09 2019)tiago_henrique_3No ratings yet
- Olhonavaga - EDITAL - VUNESP - PREFEITURA DE GUARULHOS - SP - Fiscal de Rendas - Conhecimentos GeraisDocument25 pagesOlhonavaga - EDITAL - VUNESP - PREFEITURA DE GUARULHOS - SP - Fiscal de Rendas - Conhecimentos Geraistiago_henrique_3No ratings yet
- Deus Na Natureza PDFDocument361 pagesDeus Na Natureza PDFJOSELUIZNOVELINO100% (3)
- Comunicação Na Casa EspiritaDocument72 pagesComunicação Na Casa Espiritatiago_henrique_3No ratings yet
- Lei Complementar #676, de 12 de Julho de 2016Document72 pagesLei Complementar #676, de 12 de Julho de 2016tiago_henrique_3No ratings yet
- Semântica LexicalDocument18 pagesSemântica Lexicaltiago_henrique_3No ratings yet
- Ovo em Pó - Tabela NutricionalDocument1 pageOvo em Pó - Tabela Nutricionaltiago_henrique_3No ratings yet
- Camille Flammarion - As Casas Mal AsDocument355 pagesCamille Flammarion - As Casas Mal AsMarjorie PfandeyNo ratings yet
- Manifesto - Fusão Da Engenharia Ambiental e Engenharia SanitáriaDocument3 pagesManifesto - Fusão Da Engenharia Ambiental e Engenharia Sanitáriatiago_henrique_3No ratings yet
- Camille Flammarion - Como Acabará o MundoDocument95 pagesCamille Flammarion - Como Acabará o Mundopatriciaterumi2325No ratings yet
- Como Fermentar Suas Próprias HortaliçasDocument3 pagesComo Fermentar Suas Próprias Hortaliçastiago_henrique_3No ratings yet
- Camille Flammarion - As Casas Mal AsDocument355 pagesCamille Flammarion - As Casas Mal AsMarjorie PfandeyNo ratings yet
- Camille Flammarion - Como Acabará o MundoDocument95 pagesCamille Flammarion - Como Acabará o Mundopatriciaterumi2325No ratings yet
- Deus Na Natureza PDFDocument361 pagesDeus Na Natureza PDFJOSELUIZNOVELINO100% (3)
- Camille Flammarion - Narrações Do InfinitoDocument150 pagesCamille Flammarion - Narrações Do InfinitoSergio Paulo GirardiNo ratings yet
- Camille Flammarion - O FIM DO MUNDODocument200 pagesCamille Flammarion - O FIM DO MUNDOLeonardo Barbosa100% (1)
- Camille Flammarion - UraniaDocument157 pagesCamille Flammarion - UraniaMarcelo SilvaNo ratings yet
- Manual Materias+Técnicas Tradicionais Assentamento AzulejosDocument38 pagesManual Materias+Técnicas Tradicionais Assentamento AzulejosvitorcscostaNo ratings yet
- Alternativas Energéticas - Uma Visao CemigDocument362 pagesAlternativas Energéticas - Uma Visao CemigGustavo Roscoe100% (1)
- REGULAMENTO E Normas Gerais de Uso Da Sala de Cultura 213C Atualizada em 09 09 2019Document9 pagesREGULAMENTO E Normas Gerais de Uso Da Sala de Cultura 213C Atualizada em 09 09 2019Vanessa MendesNo ratings yet
- STT507-Canal de Escoamento HidráulicoDocument27 pagesSTT507-Canal de Escoamento HidráulicoPaulo Henrique MoreiraNo ratings yet
- 0187-Química Geral PráticaDocument5 pages0187-Química Geral PráticaJuliana Mantovani de MarcoNo ratings yet
- Trabalho P536 AdDocument3 pagesTrabalho P536 AdClara SantosNo ratings yet
- Lista de Física-II-estática FluidosDocument2 pagesLista de Física-II-estática FluidosIleana MotaNo ratings yet
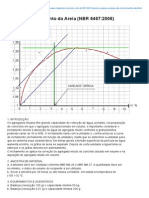
- Clube Do Concreto - Ensaio de Inchamento Da Areia (NBR 6467 - 2006)Document4 pagesClube Do Concreto - Ensaio de Inchamento Da Areia (NBR 6467 - 2006)Everton TanisNo ratings yet
- Refrigeração DomesticaDocument2 pagesRefrigeração DomesticaBruno GenteluciNo ratings yet
- Ficha de EspetrosDocument4 pagesFicha de EspetrosjoanaNo ratings yet
- Pe Ex - Revisao.av2 Ciencias 9ano 1tri PDFDocument4 pagesPe Ex - Revisao.av2 Ciencias 9ano 1tri PDFAdeptus XiaoNo ratings yet
- Aula 1 - Perspectiva Histórica Do Estudo Dos NutrientesDocument31 pagesAula 1 - Perspectiva Histórica Do Estudo Dos NutrientesFernanda Maria Vital OliveiraNo ratings yet
- C NBR 6971 - Defensas Metálicas - Projeto e ImplantaçaoDocument44 pagesC NBR 6971 - Defensas Metálicas - Projeto e ImplantaçaopaullovictorNo ratings yet
- Fisiopatologia - Tenossinovite de QuervainDocument2 pagesFisiopatologia - Tenossinovite de QuervainMateus EspindolaNo ratings yet
- Catalogo Split v21 Manaus OnepageDocument4 pagesCatalogo Split v21 Manaus OnepagemessiaslgNo ratings yet
- Trocadores de CalorDocument17 pagesTrocadores de CalorDouglas RamosNo ratings yet
- ForjamentoDocument61 pagesForjamentoWilliam CastroNo ratings yet
- Resumo FotoquímicaDocument8 pagesResumo FotoquímicaGeorge LuisNo ratings yet
- 2019-05 Bula PDFDocument12 pages2019-05 Bula PDFThiago CostaNo ratings yet
- Dnit139 2010 EsDocument8 pagesDnit139 2010 EsAlysson SantosNo ratings yet
- Exercicios Introducao A Biologia Metodo CientificoDocument5 pagesExercicios Introducao A Biologia Metodo CientificoLeidimar LustosaNo ratings yet
- Edital PPGQ 2020Document10 pagesEdital PPGQ 2020Marina PiresNo ratings yet