You might also like
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (894)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- Havells Wire Catalogue June 2021Document28 pagesHavells Wire Catalogue June 2021Lp BatNo ratings yet
- Clad-Lined Line PipeDocument21 pagesClad-Lined Line PipeAdvis100% (2)
- Waste Water TreatmentDocument3 pagesWaste Water TreatmentSana Saleem100% (1)
- CompressorDocument27 pagesCompressorsoxal100% (1)
- Investigation of Failures of 230KV Copper Conductor BushingsDocument15 pagesInvestigation of Failures of 230KV Copper Conductor BushingscalripkenNo ratings yet
- UOP PX-Plus ™ XPDocument2 pagesUOP PX-Plus ™ XPana_dcz7154No ratings yet
- Project Activity - Katherin PerezDocument1 pageProject Activity - Katherin Perezssuarez907No ratings yet
- EnglishDocument1 pageEnglishssuarez907No ratings yet
- GretaDocument1 pageGretassuarez907No ratings yet
- ParagraphDocument1 pageParagraphssuarez907No ratings yet
- EnglishDocument1 pageEnglishssuarez907No ratings yet
- WRITING - Sebastian Suarez PeñalozaDocument1 pageWRITING - Sebastian Suarez Peñalozassuarez907No ratings yet
- Relative Clauses - CompressedDocument7 pagesRelative Clauses - Compressedssuarez907No ratings yet
- EnglishDocument1 pageEnglishssuarez907No ratings yet
- Unidad3 Ingles5 PDFDocument9 pagesUnidad3 Ingles5 PDFssuarez907No ratings yet
- The Black Cat by Edgar Allan PoeDocument9 pagesThe Black Cat by Edgar Allan PoeDavid Verdugo R.No ratings yet
- The Role of Situation Awareness in Accidents of Large-Scale Technological SystemsDocument12 pagesThe Role of Situation Awareness in Accidents of Large-Scale Technological Systemsssuarez907No ratings yet
- WRITING - Sebastian Suarez PeñalozaDocument1 pageWRITING - Sebastian Suarez Peñalozassuarez907No ratings yet
- Appendices: Scenedesmus ObliquusDocument4 pagesAppendices: Scenedesmus Obliquusssuarez907No ratings yet
- NTC 1486 - NTC 5613Document197 pagesNTC 1486 - NTC 5613ssuarez907No ratings yet
- Aga RosaDocument6 pagesAga Rosassuarez907No ratings yet
- Renewable and Sustainable Energy ReviewsDocument18 pagesRenewable and Sustainable Energy Reviewsssuarez907No ratings yet
- Renewable and Sustainable Energy ReviewsDocument18 pagesRenewable and Sustainable Energy Reviewsssuarez907No ratings yet
- Li 2013Document27 pagesLi 2013ssuarez907No ratings yet
- Demostraciones Prácticas de Los Retos y Oportunidades de La Producción de Bioetanol de Primera y Segunda Generación A Partir de Cultivos TropicalesDocument6 pagesDemostraciones Prácticas de Los Retos y Oportunidades de La Producción de Bioetanol de Primera y Segunda Generación A Partir de Cultivos TropicalesBrayant Ninahuanca ParraNo ratings yet
- 12734Document21 pages12734ssuarez907No ratings yet
- 1 SR Star Jee Main GTM 02 - 03 01 2024 KeyDocument14 pages1 SR Star Jee Main GTM 02 - 03 01 2024 Keyjahnavimogarala9No ratings yet
- Problem #2b: Chromium Crystallizes With A Body-Centered Cubic Unit Cell. The Radius of ADocument8 pagesProblem #2b: Chromium Crystallizes With A Body-Centered Cubic Unit Cell. The Radius of ARadica AyuNo ratings yet
- CMB Chapter 15Document32 pagesCMB Chapter 15cyorogNo ratings yet
- Curl Activator GelDocument1 pageCurl Activator GelNemanja NikolicNo ratings yet
- Maintaining Boiler Water Quality (35 charactersDocument3 pagesMaintaining Boiler Water Quality (35 characterskcp1986No ratings yet
- Mechanical Properties For Stainless Steel FastenersDocument3 pagesMechanical Properties For Stainless Steel FastenersGonzalo MazaNo ratings yet
- Presentation 01Document28 pagesPresentation 01Rexona KhanomNo ratings yet
- B.pharm. Class NotesDocument817 pagesB.pharm. Class NotesMukesh TiwariNo ratings yet
- LP2 SCI-12 MICROBIO LABORATORY-fINALDocument21 pagesLP2 SCI-12 MICROBIO LABORATORY-fINALJOHNERROL CARCELLARNo ratings yet
- Paper 4 Jun 2001 PhysicsDocument2 pagesPaper 4 Jun 2001 Physicssolarixe100% (1)
- Chemistry, Mathematics & Physics: All India Internal Test SeriesDocument15 pagesChemistry, Mathematics & Physics: All India Internal Test Seriesmadhav aggarwalNo ratings yet
- ZN ProtocolDocument262 pagesZN ProtocolLavina D'costaNo ratings yet
- Summary KH2134 Fluid MechanicsDocument4 pagesSummary KH2134 Fluid MechanicsAzman SamerNo ratings yet
- Glass Fibre: Historical BackgroundDocument11 pagesGlass Fibre: Historical Backgroundapi-19731065100% (1)
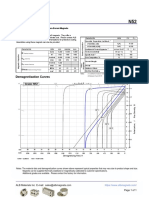
- N52 Grade Neodymium Magnets DataDocument1 pageN52 Grade Neodymium Magnets DataSteve HsuNo ratings yet
- SodaPDF-converted-Exercise No. 7 - Soil Sample Collection and PreparationDocument30 pagesSodaPDF-converted-Exercise No. 7 - Soil Sample Collection and PreparationJacky Lou GermanoNo ratings yet
- Selective Bromination With Copper (I1) Bromide - King - JOC 29 (1964)Document3 pagesSelective Bromination With Copper (I1) Bromide - King - JOC 29 (1964)dextroenantiomerNo ratings yet
- CV en - Op - Gill Giovani Awonguino oDocument1 pageCV en - Op - Gill Giovani Awonguino oAuguste SuelieNo ratings yet
- Corrosion of Iron: An Electrochemical ProcessDocument5 pagesCorrosion of Iron: An Electrochemical ProcessVickyNo ratings yet
- ANTHE 2021 (Engineering) Sample PaperDocument17 pagesANTHE 2021 (Engineering) Sample PaperDida CowernNo ratings yet
- Combustion in SI & CI EnginesDocument25 pagesCombustion in SI & CI EnginesVenkatesh KabraNo ratings yet
- AG SR SecondaryDocument33 pagesAG SR SecondaryDeepikaNo ratings yet
- Effect of Sugarmill On Soil of DoiwalaDocument11 pagesEffect of Sugarmill On Soil of DoiwalaBilal BhatNo ratings yet
- Turton - Appb 30 37Document8 pagesTurton - Appb 30 37asadNo ratings yet