You might also like
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Qualitative Research Template 1Document77 pagesQualitative Research Template 1Juliana PenadoNo ratings yet
- TOEFL Writing PDF PDFDocument40 pagesTOEFL Writing PDF PDFFaz100% (1)
- LCVP Work Experience DiaryDocument5 pagesLCVP Work Experience Diaryscolgan030967% (9)
- Chapter 1 RevisedDocument7 pagesChapter 1 RevisedChristian PalmosNo ratings yet
- Stat501.101 SU14Document2 pagesStat501.101 SU14Hafizah HalimNo ratings yet
- Gr5.Mathematics Teachers Guide JPDocument149 pagesGr5.Mathematics Teachers Guide JPRynette FerdinandezNo ratings yet
- The Effect of Mindset On Decision-MakingDocument27 pagesThe Effect of Mindset On Decision-MakingJessica PhamNo ratings yet
- 1.gilbert Et Al (Everything You Read)Document33 pages1.gilbert Et Al (Everything You Read)simiflorNo ratings yet
- Simulado InglêsDocument3 pagesSimulado InglêsJkBrNo ratings yet
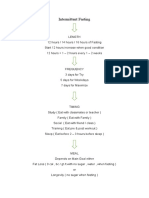
- Intermittent FastingDocument5 pagesIntermittent FastingIrfan KaSSimNo ratings yet
- Schoolcity Math q2Document2 pagesSchoolcity Math q2api-297797689No ratings yet
- GRADE 8 LP 2nd RevisionDocument3 pagesGRADE 8 LP 2nd RevisionFrezlyn ManangbaoNo ratings yet
- Britt Recommended BooksDocument6 pagesBritt Recommended Bookskamar100% (2)
- 2016 AL GIT Marking Scheme English Medium PDFDocument24 pages2016 AL GIT Marking Scheme English Medium PDFNathaneal MeththanandaNo ratings yet
- Network t1 1435 1436Document11 pagesNetwork t1 1435 1436H_BioMedNo ratings yet
- Unit 2 - Assignment Brief-đã chuyển đổiDocument5 pagesUnit 2 - Assignment Brief-đã chuyển đổiNhật HuyNo ratings yet
- The TeacherDocument51 pagesThe TeacherSonny EstrellaNo ratings yet
- Commerce BrochureDocument3 pagesCommerce BrochureSampath KumarNo ratings yet
- Literacy Unit Plan Lesson 4Document2 pagesLiteracy Unit Plan Lesson 4api-212727998No ratings yet
- Your Best Chance For: International OffersDocument9 pagesYour Best Chance For: International Offersumesh kumarNo ratings yet
- Masouda New CVDocument4 pagesMasouda New CVmasoudalatifi65No ratings yet
- Information Technology Project Management - Fifth Edition: by Jack T. Marchewka Northern Illinois UniversityDocument31 pagesInformation Technology Project Management - Fifth Edition: by Jack T. Marchewka Northern Illinois UniversitytofuNo ratings yet
- TA in PsychotherapyDocument37 pagesTA in PsychotherapyJesús RiveraNo ratings yet
- B1 Complete PackageDocument69 pagesB1 Complete PackageKhin San ThitNo ratings yet
- D'man - Mech - CTS - NSQF-5 SyllabusDocument58 pagesD'man - Mech - CTS - NSQF-5 SyllabusNitin B maskeNo ratings yet
- SpringDocument24 pagesSpringRiya IndukuriNo ratings yet
- q2 Lesson 9 Concept PaperDocument4 pagesq2 Lesson 9 Concept PaperShiela May DelacruzNo ratings yet
- Academic TextDocument2 pagesAcademic TextCatherine ProndosoNo ratings yet
- Fampulme - The Planning Functions of The ControllershipDocument22 pagesFampulme - The Planning Functions of The ControllershipCarla Pianz FampulmeNo ratings yet
- Demo K-12 Math Using 4a's Diameter of CircleDocument4 pagesDemo K-12 Math Using 4a's Diameter of CircleMichelle Alejo CortezNo ratings yet