You might also like
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5795)
- Site Accountant Store Keeper Updated On 30-11-06Document15 pagesSite Accountant Store Keeper Updated On 30-11-06farrukhsharifzadaNo ratings yet
- OLC Learning GuideDocument18 pagesOLC Learning GuidefarrukhsharifzadaNo ratings yet
- Image Segmentation Using Soft Computing: Sukhmanpreet Singh, Deepa Verma, Arun Kumar, RekhaDocument5 pagesImage Segmentation Using Soft Computing: Sukhmanpreet Singh, Deepa Verma, Arun Kumar, RekhafarrukhsharifzadaNo ratings yet
- Web Essentials: Clients, Servers, and CommunicationDocument71 pagesWeb Essentials: Clients, Servers, and CommunicationfarrukhsharifzadaNo ratings yet
- Genetic Algorithms: Machine Learning and Data MiningDocument46 pagesGenetic Algorithms: Machine Learning and Data MiningfarrukhsharifzadaNo ratings yet
- HTC Amaze 4g Tmobile UgDocument204 pagesHTC Amaze 4g Tmobile UgfarrukhsharifzadaNo ratings yet
- TestDocument1 pageTestfarrukhsharifzadaNo ratings yet
- CPS 343/543 Assignment #3 Winter Term 2005 - 2006 Due: Monday, February 13, 2006Document2 pagesCPS 343/543 Assignment #3 Winter Term 2005 - 2006 Due: Monday, February 13, 2006farrukhsharifzadaNo ratings yet
- h323 TutorialDocument30 pagesh323 TutorialfarrukhsharifzadaNo ratings yet
- Operational Semantic Meaning ForDocument2 pagesOperational Semantic Meaning ForfarrukhsharifzadaNo ratings yet
- Designing The System Architecture: Quatrani/Ch. 11 Page 139 Tuesday, November 18, 1997 5:29 PMDocument18 pagesDesigning The System Architecture: Quatrani/Ch. 11 Page 139 Tuesday, November 18, 1997 5:29 PMfarrukhsharifzadaNo ratings yet
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Quiz3 - Section001 Solution 140316Document2 pagesQuiz3 - Section001 Solution 140316kofi koomsonNo ratings yet
- AMCAT Computer Programming1 - Part2Document35 pagesAMCAT Computer Programming1 - Part2tanuj kumar100% (1)
- DSP Lab SyllabusDocument3 pagesDSP Lab SyllabusKiranKumarNo ratings yet
- What Is Naive Bayes Algorithm?Document18 pagesWhat Is Naive Bayes Algorithm?JUAN ESTEBAN LOPEZ BEDOYANo ratings yet
- Summer Workshop On Plasma Physics: Dr. Swarniv ChandraDocument17 pagesSummer Workshop On Plasma Physics: Dr. Swarniv ChandraAditi BoseNo ratings yet
- ReliabilityDocument9 pagesReliabilityWahab HassanNo ratings yet
- Multiple Choice QuestionsDocument8 pagesMultiple Choice QuestionsBan Le VanNo ratings yet
- Chapter 2 (B) - Block Ciphers and Data Encryption StandardDocument36 pagesChapter 2 (B) - Block Ciphers and Data Encryption StandardAris MunandarNo ratings yet
- Crowdnet: A Deep Convolutional Network For Dense Crowd CountingDocument5 pagesCrowdnet: A Deep Convolutional Network For Dense Crowd CountingTayyab MujahidNo ratings yet
- FINAL450Document45 pagesFINAL450BINARY BEASTNo ratings yet
- 2ND Sem Syllabus PhysicsDocument5 pages2ND Sem Syllabus PhysicsVara PrasadNo ratings yet
- American International University-Bangladesh (AIUB) Faculty of Science & Technology Course SyllabusDocument2 pagesAmerican International University-Bangladesh (AIUB) Faculty of Science & Technology Course SyllabusMahiNo ratings yet
- Control Fuzzy - PLCDocument10 pagesControl Fuzzy - PLCAlex OrozcoNo ratings yet
- Mid Point Ellipse AlgorithmDocument7 pagesMid Point Ellipse AlgorithmAman TirkeyNo ratings yet
- 030 Ice 01 Result Dec10 InsDocument41 pages030 Ice 01 Result Dec10 InsDevanshu AnandNo ratings yet
- Generalized Linear ModelDocument67 pagesGeneralized Linear Modelshanthikk3No ratings yet
- Load FlowDocument42 pagesLoad FlowTahir KedirNo ratings yet
- Ec8553 Discrete Time System ProcessingDocument1 pageEc8553 Discrete Time System Processingராஜ்குமார் கணேசன்0% (2)
- Converting A Normal Random Variable: Ask QuestionDocument4 pagesConverting A Normal Random Variable: Ask QuestionGLADY VIDALNo ratings yet
- Package Nleqslv': R Topics DocumentedDocument19 pagesPackage Nleqslv': R Topics Documentedrezza ruzuqiNo ratings yet
- A Survey On Image Data Augmentation For Deep LearnDocument49 pagesA Survey On Image Data Augmentation For Deep LearnGabriele CastaldiNo ratings yet
- Garcia, Lab 2 FinalDocument31 pagesGarcia, Lab 2 Finalaccventure5No ratings yet
- Project Plagiarism Report (Before Removing)Document21 pagesProject Plagiarism Report (Before Removing)Shyam Raj SivakumarNo ratings yet
- Differential Geometry and Mechanics Applications To Chaotic Dynamical SystemsDocument24 pagesDifferential Geometry and Mechanics Applications To Chaotic Dynamical SystemsGINOUX Jean-MarcNo ratings yet
- Richard Bird - Jeremy Gibbons - Algorithm Design With Haskell-Cambridge University Press (2020)Document454 pagesRichard Bird - Jeremy Gibbons - Algorithm Design With Haskell-Cambridge University Press (2020)scap bi100% (1)
- Online Task-Discrete ProbabilityDocument9 pagesOnline Task-Discrete ProbabilityBrenda BernardNo ratings yet
- Chapter 3 - Solving Problems by Searching Concise1Document64 pagesChapter 3 - Solving Problems by Searching Concise1SamiNo ratings yet
- SEE: Towards Semi-Supervised End-to-End Scene Text RecognitionDocument8 pagesSEE: Towards Semi-Supervised End-to-End Scene Text RecognitionOdgiiv KhNo ratings yet
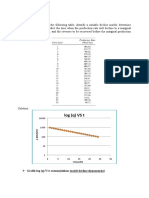
- Problems: Log (Q) VS TDocument8 pagesProblems: Log (Q) VS TDianita ZuamaNo ratings yet