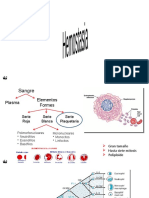
You might also like
- Prueba La Guerra de Los Duraznos 2014Document4 pagesPrueba La Guerra de Los Duraznos 2014Pamela Sepulveda Albornoz100% (1)
- Test de Ansiedad Con Variables Cuaitativas para ImprimirDocument4 pagesTest de Ansiedad Con Variables Cuaitativas para ImprimirVenicio Nerbo Davila RocanoNo ratings yet
- Acumulaciones CelularesDocument20 pagesAcumulaciones CelularesDianaNo ratings yet
- ¡Bu, Bu, Bu! La Tierra Está Llorando y Otros CuentosDocument20 pages¡Bu, Bu, Bu! La Tierra Está Llorando y Otros CuentosRoxana Hoces MontesNo ratings yet
- Salchichas Chorizos ParrillerosDocument23 pagesSalchichas Chorizos ParrillerosFreddy Omar Mora Mora100% (1)
- Introducción Al Estudio Del Desarrollo 2017 PDFDocument66 pagesIntroducción Al Estudio Del Desarrollo 2017 PDFSantiago StrizNo ratings yet
- Cuestionario TermanDocument21 pagesCuestionario TermandescargaslichaNo ratings yet
- Informe AnticoagulantesDocument27 pagesInforme AnticoagulantesJavieraPssNo ratings yet
- Factores de La CoagulaciónDocument11 pagesFactores de La Coagulaciónsweet-and-spicy_ozzy100% (1)
- Tiempo de Tromboplastina PDFDocument5 pagesTiempo de Tromboplastina PDFRoyer Dan PalmaNo ratings yet
- Anticuerpo AntiplaquetariosDocument18 pagesAnticuerpo Antiplaquetarioslourdes antonia checca escobedo100% (1)
- SindromeDocument3 pagesSindromeFernando Siul MNo ratings yet
- TransaminasasDocument32 pagesTransaminasaseveling911No ratings yet
- Evasión de patógenos al sistema de complementoDocument22 pagesEvasión de patógenos al sistema de complementoDarwin CasasNo ratings yet
- Anemias HemolíticasDocument5 pagesAnemias HemolíticasGenesis Nuñez SejolNo ratings yet
- Proteinas PlasmáticasDocument60 pagesProteinas PlasmáticasDavid Tafur Muñoz67% (6)
- 6-Sistema de ComplementoDocument9 pages6-Sistema de Complementogeobannys cedeñoNo ratings yet
- Exposicion de Estudio de La Quimiotaxis y Fagocitosis LeucocitarioDocument25 pagesExposicion de Estudio de La Quimiotaxis y Fagocitosis LeucocitarioEdinson Huamuro CastilloNo ratings yet
- Enfermedades de Las PlaquetasDocument3 pagesEnfermedades de Las PlaquetasBrayhan SánchezNo ratings yet
- Tincion de GramDocument7 pagesTincion de GramKeila NunezgutierrezNo ratings yet
- Fisiologia SanguinaDocument16 pagesFisiologia Sanguinakarlitos_escobarNo ratings yet
- Síntesis Del Grupo HemoDocument9 pagesSíntesis Del Grupo HemoDario TaimalNo ratings yet
- VSGDocument7 pagesVSGFrancisco JR Bonilla BarrerosNo ratings yet
- La Diferencia Entre Plasma y SueroDocument1 pageLa Diferencia Entre Plasma y SueroSebastian ToroNo ratings yet
- Manual de HematologiaDocument158 pagesManual de HematologiaMaioLeinenn100% (1)
- Ensayo NUEVE (Proteínas Totales Globulinas y Albúminas)Document6 pagesEnsayo NUEVE (Proteínas Totales Globulinas y Albúminas)Cbz Sebastian Zermeño BrionesNo ratings yet
- Hemoglobinuria Paroxística Nocturna (HPNDocument27 pagesHemoglobinuria Paroxística Nocturna (HPNmickevillanuevaNo ratings yet
- Sistema EndocrinoDocument3 pagesSistema Endocrinomobufe2No ratings yet
- AnticoagulantesDocument10 pagesAnticoagulantesPedro ArigatoNo ratings yet
- Práctica 4 Pruebas de CoágulaciónDocument11 pagesPráctica 4 Pruebas de CoágulaciónSandy MoralesNo ratings yet
- Cuadro Hematico CompletoDocument19 pagesCuadro Hematico CompletoyancyNo ratings yet
- Agregometria PlaquetariaDocument29 pagesAgregometria PlaquetariaKeitaro PeruNo ratings yet
- Producción de plaquetasDocument6 pagesProducción de plaquetasYuli Hernandez100% (1)
- Examen de orina: tipos de cristalesDocument26 pagesExamen de orina: tipos de cristalesCaro GuevaraNo ratings yet
- Tib Precipitación de GammaglobulinasDocument9 pagesTib Precipitación de GammaglobulinasFlavioLpNo ratings yet
- Determinación de GlucosaDocument3 pagesDeterminación de GlucosaC3b4sti4nNo ratings yet
- Proteinas TotalesDocument4 pagesProteinas TotalesAstrid Alexandra Palacio RiveraNo ratings yet
- Tiempo de Lisis de EuglobulinasDocument2 pagesTiempo de Lisis de EuglobulinasFloydRushNo ratings yet
- 3 - Fisiologia de La Sangre - Hemostasia - 2014-IDocument46 pages3 - Fisiologia de La Sangre - Hemostasia - 2014-IDharyll Chancasanampa NarvaezNo ratings yet
- Fisiopatología de Los Trastornos de La CoagulaciónDocument23 pagesFisiopatología de Los Trastornos de La CoagulaciónKelly Gabriela Lopez Ochatoma100% (1)
- Sindrome Hemorragico NeonatalDocument9 pagesSindrome Hemorragico NeonatalAdriana LuceroNo ratings yet
- Anormalidades de EritrocitosDocument58 pagesAnormalidades de EritrocitosLuis Uriel Gutierrez Diaz100% (1)
- Alteración en La Funcion PlaquetariaDocument87 pagesAlteración en La Funcion PlaquetariaJosué García FloresNo ratings yet
- 7 LeucopoyesisDocument55 pages7 LeucopoyesisFabián MoralesNo ratings yet
- Determinación de fosfatasa alcalinaDocument10 pagesDeterminación de fosfatasa alcalinaluzbnNo ratings yet
- Mecanismos de acción antimicrobianosDocument78 pagesMecanismos de acción antimicrobianosJOSE ANGEL LAZARO BARRERANo ratings yet
- Liquido AmnioticDocument49 pagesLiquido AmnioticJhon Andy Ramos100% (1)
- Desgrabacion: Sistema de ComplementoDocument12 pagesDesgrabacion: Sistema de ComplementoVeronica MendozaNo ratings yet
- Clases - Pruebas de Funcion RenalDocument36 pagesClases - Pruebas de Funcion RenalInternos de Medicina HNAL-UPSJBNo ratings yet
- Estado actual de la transfusión de hemocomponentesDocument58 pagesEstado actual de la transfusión de hemocomponentesCarlos BuitragoNo ratings yet
- (Dir. Vicente Ausina Ruiz, Santiago Moreno GuilleÌDocument692 pages(Dir. Vicente Ausina Ruiz, Santiago Moreno GuilleÌPepe DelgadoNo ratings yet
- El Sistema InmuneDocument15 pagesEl Sistema InmuneLeonela Marani NosedaNo ratings yet
- FIBRINOLISISDocument2 pagesFIBRINOLISISHellita Lv Amadeus LvNo ratings yet
- Resumen de Hemostasia SecundariaDocument5 pagesResumen de Hemostasia SecundariaSoyKlicheRodriguezNo ratings yet
- Moriremooos FusionadoDocument34 pagesMoriremooos FusionadoAri NicoleNo ratings yet
- Determinación de Fosfatasa Ácida y Fosfatasa Ácida ProstáticaDocument5 pagesDeterminación de Fosfatasa Ácida y Fosfatasa Ácida ProstáticaAngélica CruzNo ratings yet
- TrombocitopeniaDocument9 pagesTrombocitopeniaLoury Mariano SarmientoNo ratings yet
- Examen 2017 Bioquimica 4-1Document26 pagesExamen 2017 Bioquimica 4-1Daniel Moreno SalasNo ratings yet
- Anticoagulante Lúpico 2Document17 pagesAnticoagulante Lúpico 2Alison Nicol100% (1)
- Perfil hepático: guía para interpretar exámenesDocument7 pagesPerfil hepático: guía para interpretar exámenesStephanyChavezFeriaNo ratings yet
- Guia P. Bioquimica y NutricionDocument59 pagesGuia P. Bioquimica y NutricionJack Aguilar MirandaNo ratings yet
- Hemogram ADocument36 pagesHemogram Asolorockerita100% (1)
- Funciones y morfología de los leucocitosDocument2 pagesFunciones y morfología de los leucocitosLuu Moralez100% (4)
- La Cascada de CoagulaciónDocument10 pagesLa Cascada de CoagulaciónCinthia Chalaco100% (1)
- Vias de CoagulacionDocument3 pagesVias de CoagulacionAlvaro MejiaNo ratings yet
- Cascada de Coagulación y Factores de CoagulaciónDocument22 pagesCascada de Coagulación y Factores de Coagulaciónchio_hernandez100% (1)
- Fases Dela Coagulacion en La SangreDocument3 pagesFases Dela Coagulacion en La SangreaugustoNo ratings yet
- MEDICINADocument1 pageMEDICINAaugustoNo ratings yet
- Angel FibrinolisisDocument4 pagesAngel FibrinolisisaugustoNo ratings yet
- Augusto Portafolio MetabolismoDocument13 pagesAugusto Portafolio MetabolismoaugustoNo ratings yet
- Obstruccion IntestinalDocument33 pagesObstruccion Intestinaljrlol2No ratings yet
- Test Deportivos MotoresDocument13 pagesTest Deportivos MotoresViiviiana AndreaNo ratings yet
- Tarea EcogenicidadDocument12 pagesTarea EcogenicidadTaiddy MarinaNo ratings yet
- Tejido CartilaginosoDocument5 pagesTejido CartilaginosoDaniela QuirozNo ratings yet
- El AlfilerDocument2 pagesEl AlfilerkeymerNo ratings yet
- Pruebas Funcionales RenalesDocument15 pagesPruebas Funcionales Renalesrozhy_244No ratings yet
- Contabilidad AgropecuariaDocument27 pagesContabilidad AgropecuariaPaúl Irvin De La Cruz SaldañaNo ratings yet
- Epulis FisuradoDocument5 pagesEpulis FisuradoCale MillanNo ratings yet
- Microbiologia de La CarneDocument19 pagesMicrobiologia de La CarneIsidoro Neyra CamposNo ratings yet
- La mujer que mató a los pecesDocument17 pagesLa mujer que mató a los pecesJo Aqu ÍnNo ratings yet
- 10 Maneras de Utilizar El Aloe Vera o SábilaDocument6 pages10 Maneras de Utilizar El Aloe Vera o SábilaAnonymous HmRlPGINo ratings yet
- Costos de Produccion de Leche de La Cuenca Lechera de LimaDocument38 pagesCostos de Produccion de Leche de La Cuenca Lechera de LimaYEMEN MEJIA AGUIRRE100% (2)
- Enzimas SericasDocument14 pagesEnzimas Sericasmaxsd1977No ratings yet
- Pauta Ejercicios FX Platillos TibialesDocument5 pagesPauta Ejercicios FX Platillos TibialesCamila VillegasNo ratings yet
- Paleolítico vs NeolíticoDocument1 pagePaleolítico vs NeolíticoElenita Castillo HNo ratings yet
- Exposicion OsteologiaDocument30 pagesExposicion OsteologiaJuan Pa KnoNo ratings yet
- Monografia Metodos AnticonceptivosDocument27 pagesMonografia Metodos AnticonceptivosRuben JorgeNo ratings yet
- Trabajo de La Reproduccion en AnimalesDocument4 pagesTrabajo de La Reproduccion en AnimalesJuan Pablo CardonaNo ratings yet
- Guía # 6 Relaciones EcológicasDocument3 pagesGuía # 6 Relaciones EcológicasCecilia Mojica Zuleta50% (2)
- Ovario y Ciclo EstralDocument48 pagesOvario y Ciclo EstralSonia GauNo ratings yet
- 1 ConclusionDocument2 pages1 ConclusionYolima Cano SepulvedaNo ratings yet
- Acta 1 FebreroDocument6 pagesActa 1 FebreroNayibe Alejandra Pérez PeñaNo ratings yet
- 8 Animales VertebradosDocument38 pages8 Animales Vertebradosdaniela hidalgoNo ratings yet
- Corriendo (Me) Con Mi Hermana (2) Por BiffBam - TodoRelatosDocument30 pagesCorriendo (Me) Con Mi Hermana (2) Por BiffBam - TodoRelatosConejo ViejoNo ratings yet