You might also like
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- 2020 - Cat - Water Still - Purification Systems - Eng PDFDocument16 pages2020 - Cat - Water Still - Purification Systems - Eng PDFRakibul HasanNo ratings yet
- Terpenes and SteroidsDocument38 pagesTerpenes and SteroidsZabrina Tan CuaNo ratings yet
- Food Chemistry: Marinko Petrovic, Nataša Kezic, Vesna Bolanc ADocument7 pagesFood Chemistry: Marinko Petrovic, Nataša Kezic, Vesna Bolanc AIgnacio PennisiNo ratings yet
- Beta Carotene Extraction MethodDocument9 pagesBeta Carotene Extraction Methodদীপংকর রায় দীপুNo ratings yet
- Separation of Salicylic Acid Impurities With DiffeDocument4 pagesSeparation of Salicylic Acid Impurities With Diffemic92833292No ratings yet
- Jurnal Fistum 3Document8 pagesJurnal Fistum 3samhafiNo ratings yet
- User's Manual: GC1000 Mark II Process Gas ChromatographDocument77 pagesUser's Manual: GC1000 Mark II Process Gas ChromatographMohammad AbbasiNo ratings yet
- Gas Chromatography - Mass Spectrometry (Principle and Applications)Document27 pagesGas Chromatography - Mass Spectrometry (Principle and Applications)Abha GiriNo ratings yet
- 152 11 Comparison of European US Japanese Pharmacopoeia Monographs For Medicinal GasesDocument24 pages152 11 Comparison of European US Japanese Pharmacopoeia Monographs For Medicinal GasesBalesh NidhankarNo ratings yet
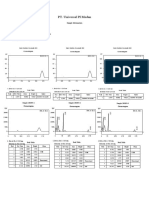
- PT. Universal PI Medan: Sample InformationDocument1 pagePT. Universal PI Medan: Sample InformationRiris HelenaNo ratings yet
- Tenofovir Disoproxil Fumarate: Riefing - Nfrared BsorptionDocument4 pagesTenofovir Disoproxil Fumarate: Riefing - Nfrared BsorptionMostofa RubalNo ratings yet
- Chromatography of Beta CaroteneDocument11 pagesChromatography of Beta CaroteneKishoNo ratings yet
- 7E Mixtures and SeparationDocument2 pages7E Mixtures and Separationakshyta gantanNo ratings yet
- Determination of Dimer in Acrylic Acid: Standard Test Methods ForDocument5 pagesDetermination of Dimer in Acrylic Acid: Standard Test Methods ForDeepak D MishraNo ratings yet
- LSD ChemistryDocument15 pagesLSD ChemistryJose Rafael Cerda CespedesNo ratings yet
- MID-1950s: ANALYSIS: Clinical Laboratory AutomationDocument15 pagesMID-1950s: ANALYSIS: Clinical Laboratory AutomationClarisse De GuzmanNo ratings yet
- Training Module: AnalysersDocument28 pagesTraining Module: Analysersmilton1987No ratings yet
- Thiamphenicol OkDocument4 pagesThiamphenicol OkJuan PerezNo ratings yet
- Dorn 2017Document13 pagesDorn 2017RamanaNo ratings yet
- Productlist 2019 21Document256 pagesProductlist 2019 21Leonardo ChNo ratings yet
- Pharm Analysis 2 - ECQ RequirementsDocument12 pagesPharm Analysis 2 - ECQ RequirementsJuliannNo ratings yet
- Biochi Ic A Et Biophysica: The Temkin Isotherm Describes Heterogeneous Protein AdsorptionDocument5 pagesBiochi Ic A Et Biophysica: The Temkin Isotherm Describes Heterogeneous Protein AdsorptionJames EdwardsNo ratings yet
- Dept. of Chem Eng. Pg-Biochemical EnggDocument57 pagesDept. of Chem Eng. Pg-Biochemical EnggH.J.PrabhuNo ratings yet
- 10.1515 Revac.2010.29.3-4.129Document20 pages10.1515 Revac.2010.29.3-4.129Snehan PeshinNo ratings yet
- Chromatography PDFDocument27 pagesChromatography PDFconstantinNo ratings yet
- Lesson Plan-Wps OfficeDocument20 pagesLesson Plan-Wps OfficeMargielyn RagosNo ratings yet
- 1124 - (Analytical Profiles of Drug Substances 7) KlausFlorey Florey (Eds.) - Academic Press (1978) PDFDocument497 pages1124 - (Analytical Profiles of Drug Substances 7) KlausFlorey Florey (Eds.) - Academic Press (1978) PDFShindi Mulfani DefaraNo ratings yet
- Supercritical Fluid Chromatography SFCDocument32 pagesSupercritical Fluid Chromatography SFCSaurab DevanandanNo ratings yet
- Analysis of SulfonatedDocument7 pagesAnalysis of SulfonatedelenitabastosNo ratings yet
- Amendment - List 03 - To - IP - 2022Document6 pagesAmendment - List 03 - To - IP - 2022QC qcNo ratings yet