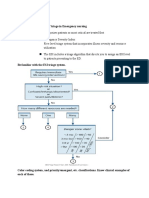
You might also like
- Sickle Cell Anemia, A Simple Guide To The Condition, Treatment And Related ConditionsFrom EverandSickle Cell Anemia, A Simple Guide To The Condition, Treatment And Related ConditionsNo ratings yet
- B HM Sickle Cell Presentation 2Document52 pagesB HM Sickle Cell Presentation 2Krupa SindhuNo ratings yet
- Sickle Cell DiseaseDocument37 pagesSickle Cell DiseaseGloria KikiNo ratings yet
- Sickle Cell DiseaseDocument23 pagesSickle Cell Diseasealejandrino_leoaugusto100% (1)
- DrepanocitozaDocument11 pagesDrepanocitozaElisa TelharajNo ratings yet
- Sickel Cell AnemiaDocument15 pagesSickel Cell AnemiarajaNo ratings yet
- Sickle Cell Anemia (HB SS) : PathogenesisDocument9 pagesSickle Cell Anemia (HB SS) : PathogenesisJennyu YuNo ratings yet
- المستند (46) كتاب الجلوةDocument11 pagesالمستند (46) كتاب الجلوةManal HassanNo ratings yet
- Untitled DocumentDocument16 pagesUntitled DocumentDinesh DoraNo ratings yet
- NCM 112 Sickle Cell AnemiaDocument6 pagesNCM 112 Sickle Cell AnemiaFifaNo ratings yet
- SCA + Thalassemia GuideDocument2 pagesSCA + Thalassemia GuideElyas MehdarNo ratings yet
- Sickle Cell Anemia: Oscass Jimmy Ruva Makchs-Bsn IiiDocument38 pagesSickle Cell Anemia: Oscass Jimmy Ruva Makchs-Bsn IiiRuva Oscass JimmyNo ratings yet
- Running Head: Sickle Cell Anemia 1Document11 pagesRunning Head: Sickle Cell Anemia 1api-264952333No ratings yet
- Sickle CellDocument7 pagesSickle Cellmahmoudmustapha93No ratings yet
- ABC: Sickle-Cell Anemia, Shock, PoisoningDocument46 pagesABC: Sickle-Cell Anemia, Shock, Poisoningroneln100% (1)
- Sickle Cell DiseaseDocument15 pagesSickle Cell DiseaseNatukunda DianahNo ratings yet
- Hemolytic AnemiaDocument23 pagesHemolytic AnemiaRashed ShatnawiNo ratings yet
- Understanding Sickle Cell AnemiaDocument10 pagesUnderstanding Sickle Cell AnemiaJulla CutaranNo ratings yet
- Sickle Cell Anaemia: The Indian PerspectiveDocument29 pagesSickle Cell Anaemia: The Indian PerspectivePadma KannanNo ratings yet
- Sickle Cell PresentationDocument4 pagesSickle Cell PresentationJoan ChoiNo ratings yet
- PallorDocument16 pagesPallorManal AlQuaimi100% (1)
- Genetic Disorders Prezzie - PPTX 12Document9 pagesGenetic Disorders Prezzie - PPTX 12RebeccaNo ratings yet
- Anemia in ChildrenDocument4 pagesAnemia in ChildrenTeslim Raji100% (1)
- Understanding Sickle Cell DiseaseDocument38 pagesUnderstanding Sickle Cell DiseasemegaNo ratings yet
- By: DR Eyad Talal: Moderator: DR I - QudaisatDocument55 pagesBy: DR Eyad Talal: Moderator: DR I - QudaisatEyad AbdeljawadNo ratings yet
- KMU Blood DisorderDocument43 pagesKMU Blood DisorderSHAFIQNo ratings yet
- Sickle Cell Anemia Term PaperDocument4 pagesSickle Cell Anemia Term Paperaflsmmmgx100% (1)
- Sickle Cell Disease, Epidemiology, Genetic History, Complication and ManagementDocument52 pagesSickle Cell Disease, Epidemiology, Genetic History, Complication and ManagementAnastasiafynnNo ratings yet
- Thalassemia: Dr. Shadab Jamal MakhdoomiDocument22 pagesThalassemia: Dr. Shadab Jamal MakhdoomiShadab JamalNo ratings yet
- NewDocument233 pagesNewRohit Gadkari100% (1)
- K-5 Pathophysiology of Cyanotic Congenital Heart DefectsDocument15 pagesK-5 Pathophysiology of Cyanotic Congenital Heart DefectsJessica GintingNo ratings yet
- Sickle Cell Anemia: By: Nancy Saber Roba Shaat Mohamed Samir El-Asaly Under Supervision: Prof. Dr. Aziza MahrousDocument43 pagesSickle Cell Anemia: By: Nancy Saber Roba Shaat Mohamed Samir El-Asaly Under Supervision: Prof. Dr. Aziza MahrousImran DogarNo ratings yet
- Sickle Cell AnemiaDocument10 pagesSickle Cell Anemiaaitzaz ul haqNo ratings yet
- Understanding Sickle Cell AnaemiaDocument18 pagesUnderstanding Sickle Cell AnaemiajhvjNo ratings yet
- MRCPCH Guide Heme: A Hypochromic MicrocyticDocument8 pagesMRCPCH Guide Heme: A Hypochromic MicrocyticRajiv KabadNo ratings yet
- Pathophysiology of Cyanotic Congenital Heart DefectsDocument16 pagesPathophysiology of Cyanotic Congenital Heart Defectsbonar46No ratings yet
- Sickle Cell Anemia - 27Document42 pagesSickle Cell Anemia - 27M.AhmedNo ratings yet
- Sickle Cell Anemia Is One of A Group of Disorders Known As Sickle Cell DiseaseDocument6 pagesSickle Cell Anemia Is One of A Group of Disorders Known As Sickle Cell DiseaseAbduladheemNo ratings yet
- Sickle Cell Anemia PDFDocument58 pagesSickle Cell Anemia PDFNithin KrishnanNo ratings yet
- Sickle Cell AnemiaDocument10 pagesSickle Cell AnemiaNader Smadi100% (1)
- Brief - Sickle Cell AnemiaDocument6 pagesBrief - Sickle Cell AnemiaJologs AggasidNo ratings yet
- Hereditary SpherocytosisDocument23 pagesHereditary SpherocytosisKashan SiddiquiNo ratings yet
- Understanding Sickle Cell AnemiaDocument68 pagesUnderstanding Sickle Cell AnemiaCAROL ANN PATITICONo ratings yet
- 12 Sickle Cell DiseaseDocument87 pages12 Sickle Cell DiseaseChristone “Zuluzulu” ZuluNo ratings yet
- Sickle Cell AnemiaDocument7 pagesSickle Cell AnemiakazelleNo ratings yet
- Sickle Cell: Kelompok 1 1. Alestya Febrimaharani 2. Iftah Shorayya 3. Lastri Sulastri 4. Siti ShopiaturohmahDocument15 pagesSickle Cell: Kelompok 1 1. Alestya Febrimaharani 2. Iftah Shorayya 3. Lastri Sulastri 4. Siti ShopiaturohmahErnesta Saulina DewiNo ratings yet
- Sickle Cell Anemia Tishya MukherjeeDocument35 pagesSickle Cell Anemia Tishya MukherjeeTishya MukherjeeNo ratings yet
- Sickle Cell AnemiaDocument15 pagesSickle Cell AnemiakavitharavNo ratings yet
- Sickle Cell DiseasesDocument13 pagesSickle Cell DiseasesValerrie NgenoNo ratings yet
- SCDDocument56 pagesSCDdrvbonillaNo ratings yet
- Sickle Cell DiseaseDocument53 pagesSickle Cell DiseaseIsaac MwangiNo ratings yet
- Sickle Cell DiseaseDocument7 pagesSickle Cell Diseasekarenkaren09100% (2)
- Sickle Cell AnemiaDocument12 pagesSickle Cell AnemiaAzwanNo ratings yet
- Sickle Cell Diseases and Thalasemia Asma2Document60 pagesSickle Cell Diseases and Thalasemia Asma2Female calmNo ratings yet
- Nursing Diagnosis and Nursing Interventions For HirschsprungDocument5 pagesNursing Diagnosis and Nursing Interventions For HirschsprungAhmed Altrafe100% (2)
- What Is Sickle Cell? Origin and Distribution of Sickle Cell DiseaseDocument27 pagesWhat Is Sickle Cell? Origin and Distribution of Sickle Cell DiseaseLuis PerazaNo ratings yet
- Hereditary Elliptocytosis: A Seminar Report OnDocument11 pagesHereditary Elliptocytosis: A Seminar Report OnSantosh SangleNo ratings yet
- Haemoglobinopathies 2Document53 pagesHaemoglobinopathies 2Igwe SolomonNo ratings yet
- SickleDocument6 pagesSickleNalini T GanesanNo ratings yet
- DEED of Abs Sale UcangDocument2 pagesDEED of Abs Sale UcangNikko SterlingNo ratings yet
- DEED of Abs Sale UcangDocument2 pagesDEED of Abs Sale UcangNikko SterlingNo ratings yet
- DebateDocument14 pagesDebateNikko SterlingNo ratings yet
- CHINA BANK CAPITAL LOSS DEDUCTIONDocument7 pagesCHINA BANK CAPITAL LOSS DEDUCTIONNikko SterlingNo ratings yet
- PALE - FinalsDocument13 pagesPALE - FinalsNikko SterlingNo ratings yet
- To Balance Its National Drug Control Program: RegulationDocument8 pagesTo Balance Its National Drug Control Program: RegulationNikko SterlingNo ratings yet
- Special Proceedings: 1. Explain The Difference Between Action and Special ProceedingDocument2 pagesSpecial Proceedings: 1. Explain The Difference Between Action and Special ProceedingNikko SterlingNo ratings yet
- Ack Bir Ampara de Leon GozonDocument2 pagesAck Bir Ampara de Leon GozonNikko SterlingNo ratings yet
- BP 22 HandluDocument8 pagesBP 22 HandluNikko SterlingNo ratings yet
- Additional Tax Cases IIDocument21 pagesAdditional Tax Cases IINikko SterlingNo ratings yet
- DepEd Butuan City job applicationDocument1 pageDepEd Butuan City job applicationNikko SterlingNo ratings yet
- 3 RR 6-2001 PDFDocument17 pages3 RR 6-2001 PDFJoyceNo ratings yet
- White Gold Marine Services VDocument4 pagesWhite Gold Marine Services VNikko SterlingNo ratings yet
- Income PDFDocument99 pagesIncome PDFNikko SterlingNo ratings yet
- The Convention On The Rights of The ChildDocument16 pagesThe Convention On The Rights of The ChildNikko SterlingNo ratings yet
- Republic Act No. 4712Document1 pageRepublic Act No. 4712Nikko SterlingNo ratings yet
- Batas Pambansa BLG 22Document3 pagesBatas Pambansa BLG 22Nikko SterlingNo ratings yet
- 2005 Conflict CasesDocument7 pages2005 Conflict CasesNikko SterlingNo ratings yet
- Bona Fide Relationship Refers To 1 A Physician and Patient Relationship Wherein A Licensed Physician Has Made A Complete Assessment of The PatientDocument4 pagesBona Fide Relationship Refers To 1 A Physician and Patient Relationship Wherein A Licensed Physician Has Made A Complete Assessment of The PatientNikko SterlingNo ratings yet
- Republic Act No. 4712Document1 pageRepublic Act No. 4712Nikko SterlingNo ratings yet
- Tax Digest 1Document2 pagesTax Digest 1Nikko SterlingNo ratings yet
- We Are Not Against Any OFW Whom We Believe and See As ModernDocument5 pagesWe Are Not Against Any OFW Whom We Believe and See As ModernNikko SterlingNo ratings yet
- TaxDocument35 pagesTaxNikko SterlingNo ratings yet
- Marubeni V CIR DigestDocument5 pagesMarubeni V CIR DigestNikko SterlingNo ratings yet
- Marine Insurance CasesDocument5 pagesMarine Insurance CasesNikko SterlingNo ratings yet
- Character 66Document1 pageCharacter 66Nikko SterlingNo ratings yet
- RR 16-2008Document8 pagesRR 16-2008Peggy SalazarNo ratings yet
- Tax Deduction Rules for Public Relations and Stock Listing FeesDocument2 pagesTax Deduction Rules for Public Relations and Stock Listing FeesNikko Sterling100% (2)
- Mendoza v. Municipality, 94 Phil. 1047 (1954) ), A Tax Levied For A Private PurposeDocument1 pageMendoza v. Municipality, 94 Phil. 1047 (1954) ), A Tax Levied For A Private PurposeNikko SterlingNo ratings yet
- Conflict DigestDocument31 pagesConflict DigestNikko SterlingNo ratings yet
- Pre Transfusion TestingDocument67 pagesPre Transfusion TestingPaulino GarciaNo ratings yet
- NCP - Activity IntoleranceDocument4 pagesNCP - Activity IntoleranceRoyce Vincent TizonNo ratings yet
- Cardiovascular SystemDocument17 pagesCardiovascular SystemPoojaNo ratings yet
- E0294Document158 pagesE0294Mayuresh DeoreNo ratings yet
- CH 19Document33 pagesCH 19Blythe Williams100% (2)
- Medonic m20 Blood Cell CounterDocument129 pagesMedonic m20 Blood Cell Counterроман жуковNo ratings yet
- SURGICAL Case ScenarioDocument6 pagesSURGICAL Case ScenarioKim Glaidyl BontuyanNo ratings yet
- Blood Defenses and FunctionsDocument18 pagesBlood Defenses and FunctionsTrúc Linh NguyễnNo ratings yet
- Mejmbps Vol11 No3 June2020 - 19Document7 pagesMejmbps Vol11 No3 June2020 - 19YUSUF SARKINGOBIRNo ratings yet
- Definitions: CLS 422 Clinical Immunohematology I Absorption and ElutionDocument6 pagesDefinitions: CLS 422 Clinical Immunohematology I Absorption and ElutionTrang HuynhNo ratings yet
- How To Check Blood Test ReportDocument3 pagesHow To Check Blood Test ReportTarkeshwar RaoNo ratings yet
- R&D Systems Hematology Products: Controls Calibrators Linearity Materials Quality Control ProgramDocument20 pagesR&D Systems Hematology Products: Controls Calibrators Linearity Materials Quality Control Programa sangNo ratings yet
- Total RBC Count by Hemocytometer - Hematology PracticalsDocument10 pagesTotal RBC Count by Hemocytometer - Hematology Practicalsrahayu_jatiningsihNo ratings yet
- RBC and WBC MorphologyDocument165 pagesRBC and WBC MorphologyZeeshan Yousuf100% (1)
- HAEMOGLOBINOPATHIESDocument111 pagesHAEMOGLOBINOPATHIESJake MillerNo ratings yet
- Blood physiology components and functionsDocument39 pagesBlood physiology components and functionsSarmad Laith MortadaNo ratings yet
- Nsg241 Study Guide Exam 5Document76 pagesNsg241 Study Guide Exam 5NatalieAndersonNo ratings yet
- Worksheet in BloodDocument12 pagesWorksheet in BloodBryan Mae H. DegorioNo ratings yet
- Hematology: Shukur Wasman Smail PHD Student in ImmunologyDocument12 pagesHematology: Shukur Wasman Smail PHD Student in ImmunologyShukr Wesman BlbasNo ratings yet
- Alvarez, Case Study CKDDocument17 pagesAlvarez, Case Study CKDErryl Justine AdvinculaNo ratings yet
- Urine Eaxmintaion ReportDocument7 pagesUrine Eaxmintaion Reportapi-3745021No ratings yet
- Professor Günther Enderlein, Doctor of Philosophy - Creator of A New Theory of Bacteria and HealthDocument6 pagesProfessor Günther Enderlein, Doctor of Philosophy - Creator of A New Theory of Bacteria and HealthSamNo ratings yet
- BREATHING & GASES EXCHANGEDocument7 pagesBREATHING & GASES EXCHANGEKankana Biswas100% (1)
- BC-6800 BrochureDocument17 pagesBC-6800 BrochureDwiyan FitraNo ratings yet
- Nclex Hema and CardioDocument9 pagesNclex Hema and CardioDefensor Pison GringgoNo ratings yet
- 6disorders of Iron Kinetics and Heme MetabolismDocument28 pages6disorders of Iron Kinetics and Heme MetabolismanonacadsNo ratings yet
- Blood ComponentsDocument17 pagesBlood ComponentsJohnSmithNo ratings yet
- Apollo Health Check Brochure Modified 2016Document14 pagesApollo Health Check Brochure Modified 2016Mahmood VdnNo ratings yet
- GRP Specimenhandbook en 2004122807Document16 pagesGRP Specimenhandbook en 2004122807AlibabarNo ratings yet
- Effectofsmokingon Red Blood Cells Count Hemoglobin Concentrationand Red CellindicesDocument5 pagesEffectofsmokingon Red Blood Cells Count Hemoglobin Concentrationand Red CellindicesIvan Matthew SuperioNo ratings yet