You might also like
- Reaction Kinetics: Reactions in SolutionFrom EverandReaction Kinetics: Reactions in SolutionRating: 3.5 out of 5 stars3.5/5 (4)
- The Transition Metals, The Lanthanides and The AntinidesDocument21 pagesThe Transition Metals, The Lanthanides and The AntinidesApril CruzNo ratings yet
- Alpha Carbon Chemistry - Enols and EnolatesDocument49 pagesAlpha Carbon Chemistry - Enols and EnolatesKuku MandavaNo ratings yet
- Practice Problems For Physical Chemistry 2Document1 pagePractice Problems For Physical Chemistry 2Fatima CellonaNo ratings yet
- Experiment 5 ChromatographyDocument3 pagesExperiment 5 ChromatographyJames Quan100% (2)
- Complex SaltDocument29 pagesComplex SaltertaNo ratings yet
- EXPERIMENT 2 Reduction of CamphorDocument2 pagesEXPERIMENT 2 Reduction of CamphorDania FaridNo ratings yet
- Effects of Caffeine and Phenobarbital on Rat Locomotor ActivityDocument5 pagesEffects of Caffeine and Phenobarbital on Rat Locomotor ActivityFeby Shyntia AfirantiNo ratings yet
- Exp 6Document8 pagesExp 6KaVisha AShaNo ratings yet
- 08 Monoglyceride eDocument14 pages08 Monoglyceride eAlf FloNo ratings yet
- Fragmentation Patterns in The Mass Spectra of Organic CompoundsDocument54 pagesFragmentation Patterns in The Mass Spectra of Organic CompoundsChandra Reddy100% (2)
- Ionic EquilibriumDocument25 pagesIonic EquilibriumTimothy James M. MadridNo ratings yet
- Chapter 3 Coordination ChemistryDocument41 pagesChapter 3 Coordination Chemistrytarun ratnaNo ratings yet
- CHEM 102 Instructional Objectives: - Additional Aqueous EquilibriaDocument29 pagesCHEM 102 Instructional Objectives: - Additional Aqueous EquilibriarajNo ratings yet
- Analytical ChemistryDocument50 pagesAnalytical ChemistryNguyễn Trịnh Anh MinhNo ratings yet
- Silly Putty Inorganic Chem LabDocument6 pagesSilly Putty Inorganic Chem LabyesbutidontuseitNo ratings yet
- Analysis of Trace Metals in Honey Using Atomic Absorption Spectroscop-Power PointDocument16 pagesAnalysis of Trace Metals in Honey Using Atomic Absorption Spectroscop-Power PointTANKO BAKONo ratings yet
- Electrochemical CellsDocument6 pagesElectrochemical Cellszeilde94% (16)
- Polymer degradation: Types & mechanismsDocument5 pagesPolymer degradation: Types & mechanismsYashi SrivastavaNo ratings yet
- Hexane and Toluene Simple and Fractional DistillationDocument12 pagesHexane and Toluene Simple and Fractional Distillationrodneyperu0% (1)
- M.SC - Chemistry SEM 2 Syllabus 2018Document14 pagesM.SC - Chemistry SEM 2 Syllabus 2018Rukam Singh TomarNo ratings yet
- Analytical Chemistry TestDocument7 pagesAnalytical Chemistry TestthecviiNo ratings yet
- Partition Coefficient DeterminationDocument4 pagesPartition Coefficient DeterminationMostafa HamawandyNo ratings yet
- Fluoride Ion Selective ElectrodeDocument14 pagesFluoride Ion Selective ElectrodeMihEugen100% (1)
- Metal Complexes or Coordination Compounds: Kfecn 4K Fe CNDocument90 pagesMetal Complexes or Coordination Compounds: Kfecn 4K Fe CNPavan Boro100% (1)
- Liquid Liquid ExtractionDocument20 pagesLiquid Liquid ExtractionGitanjali SahuNo ratings yet
- Experi Men 22Document7 pagesExperi Men 22bernardNo ratings yet
- Experiments 3 Stage 2017-2018Document50 pagesExperiments 3 Stage 2017-2018Parawgay Danar100% (1)
- Peroxo Compounds, InorganicDocument32 pagesPeroxo Compounds, InorganicKilsys AlvaradoNo ratings yet
- CHM624 Experiment (Edited Feb2015)Document14 pagesCHM624 Experiment (Edited Feb2015)Suliza SueNo ratings yet
- Objectives: FIGURE A: Example of Coordination CompoundsDocument7 pagesObjectives: FIGURE A: Example of Coordination CompoundsNurul izzatiNo ratings yet
- Theory of Indicators Ostwalds TheoryDocument3 pagesTheory of Indicators Ostwalds TheoryKala SuvarnaNo ratings yet
- 15 - Aldehyde and KetonesDocument66 pages15 - Aldehyde and KetonesIrfan Raza100% (1)
- INORGANIC CHEMISTRY EXPERIMENTSDocument46 pagesINORGANIC CHEMISTRY EXPERIMENTSpc355chyi100% (3)
- Aldol Notes PDFDocument8 pagesAldol Notes PDFAna100% (1)
- Coordination Chemistry: Invited Lectures Presented at the 20th International Conference on Coordination Chemistry, Calcutta, India, 10-14 December 1979From EverandCoordination Chemistry: Invited Lectures Presented at the 20th International Conference on Coordination Chemistry, Calcutta, India, 10-14 December 1979D. BanerjeaNo ratings yet
- Introduction to Inorganic Chemistry Lab ManualDocument23 pagesIntroduction to Inorganic Chemistry Lab Manualizz isalahNo ratings yet
- Rearrangement of Benzopinacol To Benzopinacolone TheoryDocument2 pagesRearrangement of Benzopinacol To Benzopinacolone TheoryElif YeşilyaprakNo ratings yet
- Experiment 1Document9 pagesExperiment 1Rizza Mae RaferNo ratings yet
- Anthranilic acid: precursor to tryptophanDocument20 pagesAnthranilic acid: precursor to tryptophanGlibNo ratings yet
- Note 1475054739Document10 pagesNote 1475054739Thiyaga RajanNo ratings yet
- Aromaticity CompleteDocument104 pagesAromaticity Completewahidalwahdi100% (1)
- Synthesis of TetraaminecopperDocument4 pagesSynthesis of Tetraaminecopperrahma0% (1)
- Results: Study of Reaction Kinetics: Hydrolysis of Ethyl AcetateDocument7 pagesResults: Study of Reaction Kinetics: Hydrolysis of Ethyl AcetateMuhammad Hazim TararNo ratings yet
- Stereochemistry LabDocument4 pagesStereochemistry Labmayra perezNo ratings yet
- Electrogravimetry: The Measurement of Amount of Charge Passed (Q) in Depositing The MetalDocument9 pagesElectrogravimetry: The Measurement of Amount of Charge Passed (Q) in Depositing The MetalnotmeNo ratings yet
- Experimental Measurement of Boiling Point ElevationDocument33 pagesExperimental Measurement of Boiling Point Elevationsuleman205100% (3)
- Determination of Vitamin CDocument2 pagesDetermination of Vitamin CWalwin HareNo ratings yet
- CHEMISTRY (XI, XII & Medical) by VIJAY KUMAR (M.Sc. B.Ed.)Document8 pagesCHEMISTRY (XI, XII & Medical) by VIJAY KUMAR (M.Sc. B.Ed.)Vijay Kumar100% (1)
- Determination of Inorganic Anions by Ion Chromatography PDFDocument21 pagesDetermination of Inorganic Anions by Ion Chromatography PDFJoelito MLNo ratings yet
- Carbohydrates: Color Reactions and TestsDocument19 pagesCarbohydrates: Color Reactions and TestsAjith KumarNo ratings yet
- Synthesis of Potassium Tris(oxalato)ferrate(III) TrihydrateDocument4 pagesSynthesis of Potassium Tris(oxalato)ferrate(III) Trihydratenathirahjaini0% (1)
- Inorganic Prac 2Document3 pagesInorganic Prac 2Ray DyerNo ratings yet
- EsterificationDocument5 pagesEsterificationJoonsuk LeeNo ratings yet
- Electrochemistry: Introduction To Galvanic Cells and Nernst EquationDocument3 pagesElectrochemistry: Introduction To Galvanic Cells and Nernst EquationTinuviele EsguerraNo ratings yet
- Essays on Analytical Chemistry: In Memory of Professor Anders RingbomFrom EverandEssays on Analytical Chemistry: In Memory of Professor Anders RingbomErkki WänninenNo ratings yet
- M1-M3 Biochem LecDocument36 pagesM1-M3 Biochem Lecloreign sinocruzNo ratings yet
- Ajp Module-2Document75 pagesAjp Module-2Naveen S BasandiNo ratings yet
- Section 2: Structure and Written ExpressionDocument6 pagesSection 2: Structure and Written ExpressionMalik RidwanNo ratings yet
- Lesson 1 - FractureDocument145 pagesLesson 1 - FractureMMillsNo ratings yet
- 8 PDFDocument84 pages8 PDFElijah PunzalanNo ratings yet
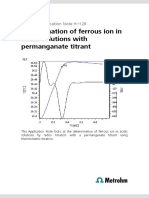
- Determination of Ferrous Ion in Acidic Solutions With Permanganate TitrantDocument3 pagesDetermination of Ferrous Ion in Acidic Solutions With Permanganate TitrantHenrique PiaggioNo ratings yet
- Estimation Solvent Activities in Polymer Solutions Using A Group-Contribution MethodDocument7 pagesEstimation Solvent Activities in Polymer Solutions Using A Group-Contribution MethodlauraNo ratings yet
- Test For LipidDocument3 pagesTest For Lipidmistic JayNo ratings yet
- TUTORIAL 1 (Additional Questions)Document2 pagesTUTORIAL 1 (Additional Questions)fatinNo ratings yet
- Yemigeba DocumentDocument78 pagesYemigeba DocumentDemelashNo ratings yet
- OilColour 14Document18 pagesOilColour 14sree kumarNo ratings yet
- Mock Test - 5Document37 pagesMock Test - 5Anirban DeNo ratings yet
- Peterson 1983Document25 pagesPeterson 1983Ryan PratamaNo ratings yet
- MANAS VIDYALAYA Practical Exams 2021-22Document9 pagesMANAS VIDYALAYA Practical Exams 2021-22Aditya RajNo ratings yet
- Portland Cement Recap AggregatesDocument127 pagesPortland Cement Recap AggregatesshayndellNo ratings yet
- All Category Name DescDocument286 pagesAll Category Name DescWeb SpringNo ratings yet
- CHEMISTRY LAB MANUAL ANSWERSDocument75 pagesCHEMISTRY LAB MANUAL ANSWERSSiddharth Rajendran75% (4)
- Nucleic Acid Extraction MethodsDocument6 pagesNucleic Acid Extraction MethodsPaul LesterNo ratings yet
- Us 5959168Document9 pagesUs 5959168RafliNo ratings yet
- II. Pericyclic Reactions: M.Sc. Semester-IV Core Course-9 (CC-9) Synthetic Organic ChemistryDocument17 pagesII. Pericyclic Reactions: M.Sc. Semester-IV Core Course-9 (CC-9) Synthetic Organic ChemistryVikasH's Digital LibraryNo ratings yet
- 001 Practice Exam 2: TTH Classes Spring 2016 Remember: Bubble in All Bubblesheet Information!Document9 pages001 Practice Exam 2: TTH Classes Spring 2016 Remember: Bubble in All Bubblesheet Information!meseret simachewNo ratings yet
- Module I-V MCQs 2 Marks 18CYB101J Virtual ExaminationDocument25 pagesModule I-V MCQs 2 Marks 18CYB101J Virtual ExaminationMAHESHWAR M R (RA2111004010136)No ratings yet
- Identifying Elements: ProblemDocument6 pagesIdentifying Elements: ProblemAdamari Andrade OrtizNo ratings yet
- General Chemistry: M. R. Naimi-JamalDocument69 pagesGeneral Chemistry: M. R. Naimi-JamalJohn Labilles Jr.No ratings yet
- Astm D664 PDFDocument11 pagesAstm D664 PDFAyoub Ghabri100% (1)
- Acid, Bases and SaltsDocument8 pagesAcid, Bases and SaltsChandanaNo ratings yet
- Sulphonation Technology in The Detergent IndustryDocument5 pagesSulphonation Technology in The Detergent IndustryСергей КубахNo ratings yet
- Rate The Quality of Your Steel - Free Webinar and Report - Learn & Share - Leica MicrosystemsDocument18 pagesRate The Quality of Your Steel - Free Webinar and Report - Learn & Share - Leica MicrosystemsharieduidNo ratings yet
- Safety Data Sheet: E6011 WELDING ELECTRODE: Information On ManufacturerDocument5 pagesSafety Data Sheet: E6011 WELDING ELECTRODE: Information On ManufacturerJennylyn DañoNo ratings yet
- "Chemical Kinetics": The Official Study Guide ForDocument21 pages"Chemical Kinetics": The Official Study Guide Foromar Alsaeed AlsaeedNo ratings yet