You might also like
- Radiation Physics and Chemistry: Traian Zaharescu, Maria Râp Ă, Eduard-Marius Lungulescu, Nicoleta Butoi TDocument10 pagesRadiation Physics and Chemistry: Traian Zaharescu, Maria Râp Ă, Eduard-Marius Lungulescu, Nicoleta Butoi TMIGUEL ANGEL GARCIA BONNo ratings yet
- Organic DyesDocument25 pagesOrganic DyesるよNo ratings yet
- Exploratory Conformational Study of Catechin JMMM 2014Document12 pagesExploratory Conformational Study of Catechin JMMM 2014AFrodita AzarNo ratings yet
- Soares Et Al For Polymer CompositesDocument16 pagesSoares Et Al For Polymer CompositesElisangelaCordeiroNo ratings yet
- Petrochemical Feedstock by Thermal Cracking of Plastic WasteDocument6 pagesPetrochemical Feedstock by Thermal Cracking of Plastic WasteWindi SetianyNo ratings yet
- Accepted Manuscript: 10.1016/j.polymertesting.2018.08.025Document25 pagesAccepted Manuscript: 10.1016/j.polymertesting.2018.08.025Mohan raj PNo ratings yet
- Kubicka Different SolventsDocument10 pagesKubicka Different SolventscligcodiNo ratings yet
- Langford LSPDocument119 pagesLangford LSPanakdamitNo ratings yet
- A Comparison Study On Non-Isothermal Decomposition Kinetics of Chitosan With Different Analysis MethodsDocument15 pagesA Comparison Study On Non-Isothermal Decomposition Kinetics of Chitosan With Different Analysis MethodsElisabeta StamateNo ratings yet
- Melt Rheology of Poly (Lactic Acid) : Entanglement and Chain Architecture AffectsDocument16 pagesMelt Rheology of Poly (Lactic Acid) : Entanglement and Chain Architecture Affectsrusheekesh3497No ratings yet
- Facile Synthesis of Precious Metal Free Ti Cu Nano Catalyst ForDocument15 pagesFacile Synthesis of Precious Metal Free Ti Cu Nano Catalyst Forcindy.medinaNo ratings yet
- 10 1021@acs Chemrev 6b00076Document238 pages10 1021@acs Chemrev 6b00076Petru Apostol100% (1)
- Liden 1988Document16 pagesLiden 1988Sajid Mohy Ul DinNo ratings yet
- C 1 JM 10649 JDocument10 pagesC 1 JM 10649 JHamada BouchinaNo ratings yet
- 1 OwalabiRasheed 23 11 2015Document14 pages1 OwalabiRasheed 23 11 2015APEX SONNo ratings yet
- O O O O O O O O O O O O O O O: Reprocessability of PHB in Extrusion: ATR-FTIR, Tensile Tests and Thermal StudiesDocument7 pagesO O O O O O O O O O O O O O O: Reprocessability of PHB in Extrusion: ATR-FTIR, Tensile Tests and Thermal StudiesJorge Gerardo Riascos QuiñonesNo ratings yet
- CatalysisDocument329 pagesCatalysisRakesh ReddyNo ratings yet
- Protonation of 5-Methylhydantoin and Its Thio Derivatives in The Gas Phase: A Theoretical StudyDocument8 pagesProtonation of 5-Methylhydantoin and Its Thio Derivatives in The Gas Phase: A Theoretical StudyProf-Zaki SafiNo ratings yet
- Modern Terpyridine Chemistry. by Ulrich S. Schubert: Randolph Thummel, University of HoustonDocument3 pagesModern Terpyridine Chemistry. by Ulrich S. Schubert: Randolph Thummel, University of HoustonAbbyNo ratings yet
- Thermal and catalytic pyrolysis of polyethyleneDocument9 pagesThermal and catalytic pyrolysis of polyethyleneKatiane MesquitaNo ratings yet
- Review Article The Thiol-Ene (Click) Reaction For The Synthesis of Plant Oil Derived PolymersDocument14 pagesReview Article The Thiol-Ene (Click) Reaction For The Synthesis of Plant Oil Derived PolymersAdnanNo ratings yet
- Control of Surface Radical Graft Polymerization OnDocument5 pagesControl of Surface Radical Graft Polymerization OnAmin ShariatmadarNo ratings yet
- Functional Group ChemistryDocument176 pagesFunctional Group ChemistryShriram Nandagopal100% (1)
- Ravindran A TH 1986Document18 pagesRavindran A TH 1986YovaAndelaSariNo ratings yet
- Water 06 01785Document22 pagesWater 06 01785ErickNo ratings yet
- Functional Group ChemistryDocument176 pagesFunctional Group Chemistrylinhmung92% (13)
- Sustainable Chemistry and Pharmacy: 2 3 Huu-Huan Pham, Sheng-Jie You, Ya-Fen Wang, Minh Thi Cao, Van-Viet PhamDocument10 pagesSustainable Chemistry and Pharmacy: 2 3 Huu-Huan Pham, Sheng-Jie You, Ya-Fen Wang, Minh Thi Cao, Van-Viet Phamsilambarasan kNo ratings yet
- Recent Advances in Olefin Metathesis and Its Application in Organic SynthesisDocument38 pagesRecent Advances in Olefin Metathesis and Its Application in Organic SynthesisTiago Breve da SilvaNo ratings yet
- Zaky Al-FatonyDocument16 pagesZaky Al-FatonyZakyAlFatonyNo ratings yet
- Synthesis and Properties of Novel Aggregation-Induced Emission Compounds With Combined Tetraphenylethylene and Dicarbazolyl Triphenylethylene MoietiesDocument9 pagesSynthesis and Properties of Novel Aggregation-Induced Emission Compounds With Combined Tetraphenylethylene and Dicarbazolyl Triphenylethylene MoietiesAsfa ChinuNo ratings yet
- N-Butane: Simulation of An Industrial Turbulent Fluidized Bed Reactor For Partial Oxidation To Maleic AnhydrideDocument10 pagesN-Butane: Simulation of An Industrial Turbulent Fluidized Bed Reactor For Partial Oxidation To Maleic AnhydrideApril JuneNo ratings yet
- Comparative Study of Lifetime Prediction Methods From Thermal Analysis of Furan Resin PDFDocument8 pagesComparative Study of Lifetime Prediction Methods From Thermal Analysis of Furan Resin PDFDos TumolvaNo ratings yet
- Thermal Stabilization of Recycled PET Through Chain Extension and Blending With PBTDocument9 pagesThermal Stabilization of Recycled PET Through Chain Extension and Blending With PBTMerve GüçlüNo ratings yet
- D H Reid - Organic Compounds of Sulphur, Selenium, and Tellurium Vol 1-Royal Society of Chemistry (1970)Document518 pagesD H Reid - Organic Compounds of Sulphur, Selenium, and Tellurium Vol 1-Royal Society of Chemistry (1970)julianpellegrini860No ratings yet
- Asm 37 PDFDocument10 pagesAsm 37 PDFJh Kurth Laura QueteguariNo ratings yet
- Literature Review of Conducting PolymersDocument6 pagesLiterature Review of Conducting Polymersmlgpufvkg100% (1)
- Thermal Degradation Behaviors of Polyethylene and PolypropyleneDocument7 pagesThermal Degradation Behaviors of Polyethylene and PolypropyleneKartik IyerNo ratings yet
- ZN - SBA-15 - Dai (2021) - Pyrolysis-Catalysis For Waste Polyolefin Conversion Into Low Aromatic NaphthaDocument13 pagesZN - SBA-15 - Dai (2021) - Pyrolysis-Catalysis For Waste Polyolefin Conversion Into Low Aromatic NaphthaKatiane MesquitaNo ratings yet
- Cross Linked PolyethyleneDocument12 pagesCross Linked PolyethyleneMichael TaylorNo ratings yet
- Oxo-Biodegradable Carbon Backbone PolymersDocument9 pagesOxo-Biodegradable Carbon Backbone PolymersDolores MasonNo ratings yet
- Estabilidade ATPDocument6 pagesEstabilidade ATPAdrian vilariño gonzalezNo ratings yet
- Recent Applications of Porphyrins As Photocatalysts in Organic Synthesis: Batch and Continuous Flow ApproachesDocument39 pagesRecent Applications of Porphyrins As Photocatalysts in Organic Synthesis: Batch and Continuous Flow Approachesareej juttNo ratings yet
- Polymer MaterialsDocument27 pagesPolymer MaterialsjawadullahNo ratings yet
- Polythiophene: From Fundamental Perspectives To ApplicationsDocument36 pagesPolythiophene: From Fundamental Perspectives To ApplicationsMarius MurgociNo ratings yet
- Novel N P Carboxy Benzyl Chitosan Poly VDocument43 pagesNovel N P Carboxy Benzyl Chitosan Poly VNafillah AbdurrahmanNo ratings yet
- University of Delhi: B.Sc. Industrial ChemistryDocument80 pagesUniversity of Delhi: B.Sc. Industrial ChemistrygaursandeepNo ratings yet
- Phase Transfer Catalysis Chemistry and Engineering - Journal Review - AIChE, Mar 1998, 44 (3), 612Document35 pagesPhase Transfer Catalysis Chemistry and Engineering - Journal Review - AIChE, Mar 1998, 44 (3), 612muopioidreceptorNo ratings yet
- Paper 2011 ISIDocument5 pagesPaper 2011 ISISayed ShaarawiNo ratings yet
- Catalytic Conversion of Polyolefins Into Liquid Fuels Over Mcm-41: Comparison With Zsm-5 and Amorphous Sio Al ODocument7 pagesCatalytic Conversion of Polyolefins Into Liquid Fuels Over Mcm-41: Comparison With Zsm-5 and Amorphous Sio Al OZahid FarooqNo ratings yet
- BK9780854041862 00001Document4 pagesBK9780854041862 00001Chrstina Adel TawfiqNo ratings yet
- Chemical Kinetics of Biomass Pyrolysis: Energy & Fuels January 2009Document11 pagesChemical Kinetics of Biomass Pyrolysis: Energy & Fuels January 2009Claudia MendozaNo ratings yet
- Reaction Mechanism of Soot Formation in AmesDocument10 pagesReaction Mechanism of Soot Formation in AmesZavaleta RedlerNo ratings yet
- Journal of Catalysis: SciencedirectDocument8 pagesJournal of Catalysis: SciencedirectLerchundi KityNo ratings yet
- 2019 - AEM - PCE11.o-IDTBR-MW and Miscibility Correlating With Device Performance and MorphologyDocument14 pages2019 - AEM - PCE11.o-IDTBR-MW and Miscibility Correlating With Device Performance and MorphologyBilal NaveedNo ratings yet
- Sustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimeFrom EverandSustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimeNo ratings yet
- Handbook of Oligo- and PolythiophenesFrom EverandHandbook of Oligo- and PolythiophenesDenis FichouNo ratings yet
- Application of Nuclear Magnetic Resonance Spectroscopy in Organic Chemistry: International Series in Organic ChemistryFrom EverandApplication of Nuclear Magnetic Resonance Spectroscopy in Organic Chemistry: International Series in Organic ChemistryRating: 1 out of 5 stars1/5 (1)
- Progress in Physical Organic ChemistryFrom EverandProgress in Physical Organic ChemistryRobert W. TaftNo ratings yet
- Organotransition Metal Chemistry: Applications to Organic Synthesis: Applications to Organic SynthesisFrom EverandOrganotransition Metal Chemistry: Applications to Organic Synthesis: Applications to Organic SynthesisNo ratings yet
- Organometallic Chemistry: Plenary Lectures Presented at the Eighth International Conference on Organometallic Chemistry, Kyoto, Japan, 12-16 September 1977From EverandOrganometallic Chemistry: Plenary Lectures Presented at the Eighth International Conference on Organometallic Chemistry, Kyoto, Japan, 12-16 September 1977Y. IshiiNo ratings yet
- Technical Information: TMS - VHPDocument1 pageTechnical Information: TMS - VHPVictor CastrejonNo ratings yet
- Technical Information: Pyrobloc® Sap2 and Sap5Document2 pagesTechnical Information: Pyrobloc® Sap2 and Sap5Victor CastrejonNo ratings yet
- Polymer additive for polyester productionDocument1 pagePolymer additive for polyester productionVictor CastrejonNo ratings yet
- Pyrobloc SAP-2 PDFDocument1 pagePyrobloc SAP-2 PDFVictor CastrejonNo ratings yet
- Technical Information: Microfine VHPDocument1 pageTechnical Information: Microfine VHPVictor CastrejonNo ratings yet
- Tmse PDFDocument1 pageTmse PDFVictor CastrejonNo ratings yet
- Pyrobloc SAP-5 PDFDocument1 pagePyrobloc SAP-5 PDFVictor CastrejonNo ratings yet
- Technical Information: Sodium Antimonate-EDocument1 pageTechnical Information: Sodium Antimonate-EVictor CastrejonNo ratings yet
- Suptms PDFDocument1 pageSuptms PDFVictor CastrejonNo ratings yet
- Technical Information: TMS Catalyst Grade PHPDocument1 pageTechnical Information: TMS Catalyst Grade PHPVictor CastrejonNo ratings yet
- Technical Information: Antimony Trioxide Wetted GradesDocument1 pageTechnical Information: Antimony Trioxide Wetted GradesVictor CastrejonNo ratings yet
- Atstemp PDFDocument1 pageAtstemp PDFVictor CastrejonNo ratings yet
- Tmse PDFDocument1 pageTmse PDFVictor CastrejonNo ratings yet
- Tms PDFDocument2 pagesTms PDFVictor CastrejonNo ratings yet
- Ongard 2 PDFDocument1 pageOngard 2 PDFVictor CastrejonNo ratings yet
- Technical Information: Antimony MetalDocument1 pageTechnical Information: Antimony MetalVictor CastrejonNo ratings yet
- Technical Information: Microfine Ao5 and Ao5-Hp Antimony TrioxideDocument2 pagesTechnical Information: Microfine Ao5 and Ao5-Hp Antimony TrioxideVictor CastrejonNo ratings yet
- Technical Information: Atc-HclDocument1 pageTechnical Information: Atc-HclVictor CastrejonNo ratings yet
- Technical Information: Microfine Ao5 and Ao5-Hp Antimony TrioxideDocument2 pagesTechnical Information: Microfine Ao5 and Ao5-Hp Antimony TrioxideVictor CastrejonNo ratings yet
- Atomao9 PDFDocument2 pagesAtomao9 PDFVictor CastrejonNo ratings yet
- Polymer One competitive step aheadDocument1 pagePolymer One competitive step aheadVictor CastrejonNo ratings yet
- Technical Information: Antimony TrichlorideDocument1 pageTechnical Information: Antimony TrichlorideVictor CastrejonNo ratings yet
- Technical Information: Microfine Catalyst Grade PVHP Antimony TrioxideDocument1 pageTechnical Information: Microfine Catalyst Grade PVHP Antimony TrioxideVictor CastrejonNo ratings yet
- Atocg PDFDocument1 pageAtocg PDFVictor CastrejonNo ratings yet
- PolymerAdditives - Thanox 1076 - TDS - 2012-JuneDocument1 pagePolymerAdditives - Thanox 1076 - TDS - 2012-JuneVictor CastrejonNo ratings yet
- PolymerAdditives - Thanox 1098 - TDS - April-2013Document1 pagePolymerAdditives - Thanox 1098 - TDS - April-2013Victor CastrejonNo ratings yet
- Technical Information: Microfine Catalyst Grade PHP Antimony TrioxideDocument1 pageTechnical Information: Microfine Catalyst Grade PHP Antimony TrioxideVictor CastrejonNo ratings yet
- Atomeg PDFDocument2 pagesAtomeg PDFVictor CastrejonNo ratings yet
- PolymerAdditives - Thanox 1010 - TDS - 2011-AugustDocument1 pagePolymerAdditives - Thanox 1010 - TDS - 2011-AugustVictor CastrejonNo ratings yet
- THANOX 1035 Technical Data SheetDocument1 pageTHANOX 1035 Technical Data SheetVictor CastrejonNo ratings yet
- Chapter 6 Exercises (Bonds & Interest)Document2 pagesChapter 6 Exercises (Bonds & Interest)Shaheera SuhaimiNo ratings yet
- Tabel Benkelman Beam Baru - AsisDocument21 pagesTabel Benkelman Beam Baru - AsisAsisNo ratings yet
- The Indonesian Food Processing Industry (Final)Document48 pagesThe Indonesian Food Processing Industry (Final)patalnoNo ratings yet
- Minicap FTC260, FTC262: Technical InformationDocument20 pagesMinicap FTC260, FTC262: Technical InformationAmanda PorterNo ratings yet
- RepairManual NEF M100 M150 P3D32N003E Mar06Document172 pagesRepairManual NEF M100 M150 P3D32N003E Mar06manuel segovia100% (1)
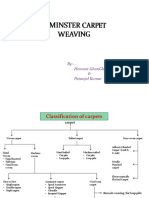
- Axminster CarpetDocument19 pagesAxminster Carpetrohit sinhaNo ratings yet
- 1 5109354204116287644 PDFDocument336 pages1 5109354204116287644 PDFGerardoNo ratings yet
- AREADocument10 pagesAREAhaipm1979No ratings yet
- Crusher Type Part Number Description Weight (KG)Document3 pagesCrusher Type Part Number Description Weight (KG)Juvenal CorreiaNo ratings yet
- Fakeaway - Healthy Home-Cooked Takeaway MealsDocument194 pagesFakeaway - Healthy Home-Cooked Takeaway MealsBiên Nguyễn HữuNo ratings yet
- Descent of Darwin - Theosophy WatchDocument17 pagesDescent of Darwin - Theosophy Watchjorge_lazaro_6No ratings yet
- Rom BlonDocument8 pagesRom BlonCharlesJustin AyunonNo ratings yet
- ABRACON's Tuning Fork Crystals and Oscillators for 32.768kHz RTC ApplicationsDocument13 pagesABRACON's Tuning Fork Crystals and Oscillators for 32.768kHz RTC Applicationsdit277No ratings yet
- PV Elite ResultDocument18 pagesPV Elite ResultVeny MartianiNo ratings yet
- Strategic Capacity ManagementDocument36 pagesStrategic Capacity ManagementRahul KhannaNo ratings yet
- Laplace Transform AssignmentDocument1 pageLaplace Transform AssignmentMohamad DuhokiNo ratings yet
- Dr. Kumar's Probability and Statistics LectureDocument104 pagesDr. Kumar's Probability and Statistics LectureAnish KumarNo ratings yet
- Zip Grade 100 Question V2Document1 pageZip Grade 100 Question V2Jesus Daniel Anaya AlvaradoNo ratings yet
- RDT Steering System Pressure ChartDocument118 pagesRDT Steering System Pressure ChartAnonymous 340A7vnwV1100% (2)
- 6013 GCS-CONTROLS enDocument5 pages6013 GCS-CONTROLS enMuhammad SyaqirinNo ratings yet
- 21734Document67 pages21734Jeef100% (4)
- CQ B TECHNIQUESDocument37 pagesCQ B TECHNIQUESeddie6355100% (3)
- 2G - 4G Network Module - Data Sheet - EnglishDocument8 pages2G - 4G Network Module - Data Sheet - EnglishbbwroNo ratings yet
- ADAMHAND8A4Document11 pagesADAMHAND8A4Elker José Camargo100% (1)
- Network FYPDocument3 pagesNetwork FYPla tahzanNo ratings yet
- 2G Call FlowDocument71 pages2G Call Flowm191084No ratings yet
- CP I-O Modules PDFDocument91 pagesCP I-O Modules PDFVlad ChioreanNo ratings yet
- Contact Details For Medical Schools by PostcodeDocument13 pagesContact Details For Medical Schools by PostcodeHeena R ModiNo ratings yet
- Yamaha TT600RE Service ManualDocument382 pagesYamaha TT600RE Service ManualStefan30393% (14)